8.2.3
Opioid analgesic therapy
Geoffrey Hanks
Nathan I. Cherny
Marie Fallon
Introduction
Treatment with analgesic drugs is the mainstay of cancer pain management.(1,2)
Although concurrent use of other approaches and interventions may be
appropriate in many patients, and necessary in some, analgesic drugs
are needed in almost every case. Drugs whose primary clinical action is
the relief of pain are conventionally classified on the basis of their
activity at opioid receptors as either opioid or non-opioid analgesics.
A third class, adjuvant analgesics, are drugs with other primary
indications that can be effective analgesics in specific circumstances.
The major group of drugs used in cancer pain management are the opioid
analgesics.
During the last 20 years there has been a dramatic
increase in our knowledge of the sites and mechanism of action of the
opioids. The development of analytical methods has also been of great
importance in facilitating pharmacokinetic studies of the disposition
and fate of opioids in patients. More recently advances in genomic
research have indicated the potential importance of pharmacogenetic
factors in the response to opioid analgesics.(3)
These studies have begun to offer us a better understanding of some of
the sources of variation between individuals in their response to
opioids and to suggest ways of minimizing some of their adverse
effects. Although there are gaps in our knowledge of opioid
pharmacology, the rational and appropriate use of these drugs is based
on the knowledge of their pharmacological properties derived from
well-controlled clinical trials.
Terminology
In this chapter and throughout this text, we have adopted the following conventions in terminology.
Opiate is a specific term
that is used to describe drugs derived from the juice of the opium
poppy. For example, morphine is an opiate but methadone (a completely
synthetic drug) is not.(4)
Opioid is a general term
that includes naturally occurring, semi-synthetic, and synthetic drugs
which produce their effects by combining with opioid receptors and are
stereospecifically antagonized by naloxone. In this context we refer to
opioid agonists, opioid antagonists, opioid peptides, and opioid
receptors.
Narcotic is commonly used to describe morphine-like drugs and other drugs of abuse. The term is derived from the Greek narke,
meaning numbness or torpor. Since this is an imprecise and pejorative
term that is not useful in a pharmacological context, its use with
reference to opioids is discouraged. The term narcotic is not used in
this book.
Opioid receptors
Opioids are agonists at highly specific receptor sites,
and there is general agreement on the existence of at least three types
of opioid receptor: the morphine receptor µ (mu), the κ (kappa)
receptor at which the prototype agonist is ketocyclazocine, and the
enkephalin receptor δ (delta). A fourth receptor, σ (sigma), was
originally included in this group. However, actions mediated through σ
receptors are not reversed by naloxone so it is not a true opioid
receptor. The µ receptors have been further sub-classified into two
distinct sub-types (µ1 and µ2), as have the δ receptors (δ1 and δ2).
Kappa receptors have been divided into κ1, κ2, and κ3 sub-types.
Recently, several of these receptors have been successfully cloned.
In animal models, some laboratories have cloned up to 10µ receptor sub-types.(5) However, the functional significance of these ‘spliced variants’ remains unclear at present
Table 1 shows the putative effects mediated by the three main opioid receptors.(6) This classification is based on the original description by Martin et al.(7)
The effects presumed to be mediated at µ receptors have been defined as
a result of both human and animal studies, while the effects mediated
at κ receptors derive predominantly from animal models. κ receptors
mediate analgesia that persists in animals made tolerant to δ agonists;
κ agonists produce less respiratory depression and miosis than µ
agonists. It is assumed that opioid receptors mediate the sedative and
mental clouding effects of opioids, in addition to their other
pharmacological actions.
Table 1 Responses mediated by activation of opioid receptors | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
P.317
Opioid receptors are found in several areas of the
brain, particularly in the periaqueductal grey matter, and throughout
the spinal cord. Supra-spinal systems have been described for µ1, κ3,
and δ2 receptors, whereas µ2, κ1, and δ1 receptors modulate pain at the
spinal level.(8) Our understanding of the
effect profiles of opioid receptors remains incomplete, as new advances
make it clear that their disposition and structure are extremely
complex.
The molecular pharmacology of opioids
Molecular biology techniques have enabled the primary
amino acid sequence of the human µ,κ, and δ opioid receptors to be
determined. The pharmacological and functional properties of the cloned
receptors, the development of ‘knockout’ animals (which are deficient
in a receptor or part of a receptor), and the manipulation and
substitution of various amino acids in critical domains of the various
opioid receptors are providing new information on receptor function and
organization that will lead to an increased understanding of opioid
neurotransmission at the molecular level and of the factors controlling
the development of responses to opioid drugs.
The three opioid receptor genes, encoding mu (MOR),
delta (DOR), and kappa (KOR) have been cloned. The binding affinities
of a range of opioids to the µ-, κ-, and δ-opioid receptors and also to
the cloned µ receptor have been examined in animals. The animal data
indicate that while the commonly prescribed opioids (agonists and
antagonists) bind preferentially to the µ receptor, they also interact
with all three receptor types. Morphine and normorphine (a minor
metabolite of morphine) show the greatest relative preference for the µ
receptor. Methadone (which also has some NMDA-receptor blocking
activity) shows significant binding to δ receptors, while
buprenorphine, and to a lesser extent naloxone, avidly bind to all
three receptor types. There is evidence (albeit inconsistent) that the
D-enantiomer of methadone blocks the NMDA receptor. The binding
affinity of buprenorphine to the µ receptor is much greater than that
of naloxone, which explains why the latter only partially reverses
buprenorphine toxicity.
Animal data also indicate that codeine and diamorphine
have very poor binding to opioid receptors, which reinforces the
possibility that both are prodrugs where the pharmacologically active
species are morphine(9) and 6-monoacetyl morphine,(10)
respectively. Oxycodone may also act through an active metabolite,
though there are some data which suggest that this is not the case.(11)
Pethidine is considered to be a potent µ receptor
agonist, but it does bind weakly to all three opioid receptors.
Ketobemidone has a lower affinity for the µ receptor than does
morphine, but it shows greater discrimination for this receptor
compared to κ receptors. The binding of both of these opioids to the δ
receptor is similar.(12)
The binding of morphine, methadone, buprenorphine, and
naloxone to the cloned human µ receptor shows excellent congruence with
the animal data.(13,14)
Fentanyl shows a similar binding affinity, while codeine demonstrates
greater binding affinity to the cloned human receptor. Thus, for these
commonly administered opioids, there is no great variability in their
affinity for the human µ receptor.
The clinical relevance of these data is that different
opioids act in different ways. We are aware too from anecdotal clinical
experience that there is considerable interindividual variability in
response to each opioid and this reinforces our need to assess an
individual's response to opioid analgesia carefully. It would be
premature to extrapolate from laboratory data, which in many instances
have not yet been replicated, to the clinic. However, these data
increasingly inform the clinical use of these drugs and will be
particularly relevant to new approachs to their use such as ‘opioid
switching’.
Agonists, antagonists, potency, and efficacy
Based on their interactions with the various receptor
sub-types, opioid compounds can be divided into agonist,
agonist–antagonist, and antagonist classes (Table 2).
Table 2 Classification of opioid analgesics into agonist, agonist–antagonist and antagonist classes | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Agonists
An agonist is a drug that has affinity for and binds to
cell receptors to induce changes in the cell that stimulate
physiological activity. The agonist opioid drugs have no clinically
relevant ceiling effect to analgesia. As the dose is raised, analgesic
effects increase in a log linear function, until either analgesia is
achieved or dose-limiting adverse effects supervene. Efficacy is
defined by the maximal response induced by administration of the active
agent. In practice, this is determined by the degree of analgesia
produced following dose escalation through a range limited by the
development of adverse effects. Potency, in contrast, reflects the
dose–response relationship. Potency is influenced by pharmacokinetic
factors (i.e. how much of the drug enters the body systemic circulation
and then reaches the receptors) and by affinity to drug receptors.
The concepts of efficacy and potency are illustrated in Fig. 1,
which shows the dose–response curves for two drugs A and B. If the
logarithm of dose is plotted against response an agonist will produce
an S-shaped or sigmoid curve. The efficacy of the two drugs, defined by
maximum response is the same. Drug A produces the same response as B
but at a lower dose, and therefore is described as more potent.
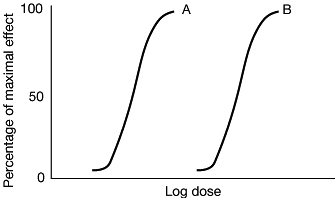 |
Fig. 1
Dose-response curves for two full opioid agonists (A and B) similar in
efficacy but different in potency (A is more potent than B). |
Antagonist
Antagonist drugs have no intrinsic pharmacological
action but can interfere with the action of an agonist. Competitive
antagonists bind to the same receptor and compete for receptor sites,
whereas non-competitive antagonists block the effects of the agonist in
some other way.
Agonist–antagonist
The agonist–antagonist analgesics can, in turn, be
subdivided into the mixed agonist–antagonists and the partial agonists,
a distinction also based
on specific patterns of drug–receptor interaction. Both the partial agonist and agonist–antagonist drugs have a ceiling effect for analgesia, and although they produce analgesia in the opioid-naive patient, in theory they can precipitate withdrawal in patients who are physically dependent on morphine-like drugs. For these reasons, they have been considered generally to have a limited role in the management of patients with cancer pain.
P.318
on specific patterns of drug–receptor interaction. Both the partial agonist and agonist–antagonist drugs have a ceiling effect for analgesia, and although they produce analgesia in the opioid-naive patient, in theory they can precipitate withdrawal in patients who are physically dependent on morphine-like drugs. For these reasons, they have been considered generally to have a limited role in the management of patients with cancer pain.
Mixed agonist–antagonists
The mixed agonist–antagonist drugs produce agonist
effects at one receptor and antagonist effects at another. Pentazocine
is the prototype agonist- antagonist: it has agonist effects at κ
receptors and weak µ antagonist actions. Thus in addition to analgesia,
pentazocine may produce κ-mediated psychotomimetic effects not seen
with full or partial µ agonists. When a mixed agonist–antagonist is
administered together with an agonist, the antagonist effect at the µ
receptor can generate an acute withdrawal syndrome.
Partial agonists
A partial agonist has low intrinsic activity (efficacy)
so that its dose- response curve exhibits a ceiling effect at less than
the maximum effect produced by a full agonist. Buprenorphine is the
main example of a partial agonist opioid. Increasing the dose of such a
drug above its ceiling does not result in any further increase in
response. This phenomenon is illustrated in Fig. 2
in which C is a partial agonist. C is more potent than B (in the lower
part of the curve it will produce the same response at a lower dose),
but is less effective than both A and B because of its ceiling effect.
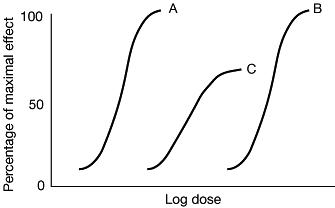 |
Fig. 2 Dose-response curves for two full opioid agonists (A and B) and a partial opioid agonist C. |
When a partial agonist is administered together with an
agonist, displacement of the agonist can cause a net reduction in
pharmacological action which may be sufficient to generate an acute
withdrawal syndrome. Whilst this is a theoretical possibility with
morphine and buprenorphine, no such interaction has been reported.
Similarly, it has been suggested that the effects of morphine may be
blocked in a patient switched from buprenorphine, because of the
prolonged action of buprenorphine and the assumption that it will
‘antagonize’ the effect of morphine. This has been one of the reasons
why buprenorphine has not been in cancer pain management. However, the
recent development of a transdermal formulation of buprenorphine may
encourage its use in chronic cancer pain (and chronic non-cancer pain).
An analgesic ceiling with buprenorphine is only reached at doses of
8–16 mg or more in 24 h.(15) When used in usual
recommended doses (for example, two patches of 70 µg/h of transdermal
buprenorphine, equivalent to 3–4 mg per 24 h) buprenorphine can be
considered a full µ agonist since at these doses its effect will lie on
the linear part of the dose–response curve.
Relative potency and equianalgesic doses
Relative potency is the ratio of the doses of two
analgesics required to produce the same analgesic effect. By convention
the relative potency of each of the commonly used opioids is based upon
a comparison with 10 mg of parenteral morphine. Data from single- and
repeated-dose studies in patients with acute or chronic pain have been
used to develop an equianalgesic dose table (Table 3)
that provides guidelines for dose selection when the drug or route of
administration is changed. The information contained in the
equianalgesic dose table does not represent standard doses, nor is it
intended as an absolute guideline for dose selection. Many variables
may influence the appropriate dose for an individual patient, including
intensity of pain, prior opioid exposure in terms of drug, duration,
and dose (and the degree of cross-tolerance that this confers), age,
route of administration, level of consciousness, metabolic
abnormalities (see below), and genetic polymorphism in the expression
of relevant enzymes or receptors.
Table 3 Opioid analgesics (pure µ agonists) used for the treatment of chronic pain | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Dose–response relationship
As noted above, there is no ceiling to the analgesic
effects of full agonist opioids. As the dose is raised, analgesic
effects increase as a log linear function. In practice, the appearance
of adverse effects, including confusion, sedation, nausea, vomiting, or
respiratory depression, imposes a limit on the useful dose of an opioid
agonist. Thus the efficacy of any particular drug in an individual
patient will be determined by the degree of analgesia produced
following dose escalation to intolerable and unmanageable side-effects.
The role of opioids in the management of cancer pain
Analgesic therapy with opioids, non-opioids, and
adjuvant analgesics is developed for the individual patient through a
process of continuous evaluation so that a favourable balance between
pain relief and adverse pharmacological effects is maintained.
The analgesic ladder
An expert committee convened by the Cancer and
Palliative Care Unit of the World Health Organization (WHO) proposed a
structured approach to drug selection for cancer pain, which has become
known as the ‘WHO analgesic ladder’.(16) When
combined with appropriate dosing guidelines, this approach is capable
of providing adequate relief to 70–90 per cent of patients.(17,18,19,20,21,22 and 23)
Emphasizing that the intensity of pain, rather than its specific
aetiology, should be the prime consideration in analgesic selection,
the approach advocates three basic steps (Fig. 3):
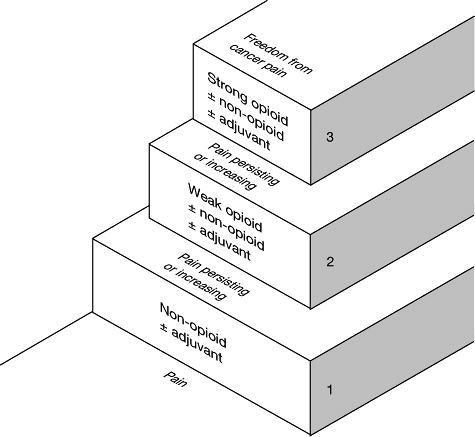 |
Fig. 3 The WHO three-step analgesic ladder. (Reproduced with permission from ref. 2.) |
- Patients with mild cancer-related pain should be treated with a non-opioid analgesic, which should be combined with adjuvant drugsP.319
P.320
P.321
if a specific indication for these exists. For example, a patient with mild to moderate arm pain caused by radiation-induced brachial plexopathy may benefit when a tricyclic antidepressant is added to paracetamol (acetaminophen).(24,25) - Patients who are relatively non-tolerant and present with moderate pain, or who fail to achieve adequate relief after a trial of a non-opioid analgesic, should be treated with an opioid conventionally used for mild to moderate pain (formerly known as a ‘weak’ opioid). This treatment is typically accomplished using a combination product containing a non-opioid (e.g. aspirin or paracetamol) and an opioid (such as codeine, oxycodone, or propoxyphene). This combination can also be coadministered with an adjuvant analgesic. The doses of these combination products can be increased until the maximum dose of the non-opioid analgesic is attained (e.g. 4000–6000 mg paracetamol); beyond this dose, the opioid contained in the combination product could be increased as a single agent, or the patient could be switched to an opioid conventionally used in step 3.
- Patients who present with severe pain, or who fail to achieve adequate relief following appropriate administration of drugs on the second step of the analgesic ladder, should receive an opioid conventionally used for moderate to severe pain (formerly known as a ‘strong’ opioid). This group includes morphine, diamorphine, fentanyl, oxycodone, phenazocine, hydromorphone, methadone, levorphanol, and oxymorphone. These drugs may also be combined with a non-opioid analgesic or an adjuvant drug. Clearly, the boundary between opioids used in the second and third steps of the analgesic ladder is somewhat artificial since low doses of morphine or other opioids for severe pain can be less effective than high doses of codeine or propoxyphene.
According to these guidelines, a trial of opioid therapy
should be given to all patients with pain of moderate or greater
severity.
The evidence of the long-term efficacy of this approach
and the evidence base underlying its recommendations has been the
subject of criticism.(26) Several other
developments have also contributed to a reevaluation of the WHO ladder.
The introduction of low-dose formulations of opioid agonists
traditionally used for severe pain, and of other agents such as
tramadol has widened the repertoire of agents suitable for the
management of moderate pain (step 2). Indeed, many authorities now
advocate the use of the same opioid for all pains of moderate or
greater intensity.(27,28,29 and 30)
Despite these reservations, the guiding principle that
analgesic selection should be primarily determined by the severity of
the pain remains sound, and continues to be widely endorsed.(1,27,31)
Opioid analgesics
The division of opioid agonists into ‘weak’ or ‘strong’
opioids, which was incorporated into the original analgesic ladder
proposed by the WHO, was not based on fundamental differences in their
pharmacology, but rather reflected the customary manner in which these
drugs were used. In this chapter we will refer to opioids for mild to
moderate pain and opioids for moderate to severe pain rather than
‘weak’ or ‘strong’ opioids. This terminology is now incorporated into
the current version of the WHO analgesic ladder.
Opioids for mild to moderate pain
Codeine
Codeine (methylmorphine) is a naturally occurring opium alkaloid used as an analgesic, antitussive, and antidiarrhoeal agent (Fig. 4).
Codeine is much less potent than morphine and produces its analgesic
effects in part by binding to µ opioid receptors but with low affinity
and, in part through biotransformation to morphine by cytochrome P-450
CYP2D6 (sparteine oxygenase) which exhibits genetic polymorphism.
Approximately 7 per cent of Caucasians lack CYP2D6 activity (poor
metabolizers) due to inheritance of two non-functional alleles and in
these individuals codeine has a diminished analgesic effect.(32,33)
 |
Fig. 4 The chemical structures of morphine and codeine. |
Codeine phosphate is absorbed well from the
gastrointestinal tract, but oral bioavailability varies considerably
between individuals (from 12 to 84 per cent in one study(34)).
The main metabolite is codeine-6-glucuronide, with much smaller amounts
of norcodeine, morphine, and morphine 3- and 6-glucuronides also being
produced.(35) The usual oral dose of codeine is 30–60 mg and its duration of action is 4–6 h.
Codeine is not generally given as a single agent when
used orally as an analgesic, but is usually combined with a non-opioid
and recent systematic reviews confirm that the combination of codeine
and paracetamol is more effective that paracetamol alone.(36,37)
A sustained-release formulation of codeine is available in some
countries. When changing from regular administration of a
codeine/non-opioid combination to morphine, patients receiving a total
daily dose of 240–360 mg codeine are usually started on 60 mg morphine
daily.
Dihydrocodeine
Dihydrocodeine is a semi-synthetic analogue of codeine
that is used as an analgesic, antitussive, and antidiarrhoeal agent.
When administered by mouth, dihydrocodeine is equianalgesic to codeine.
However, when administed parenterally it is approximately twice as
potent as codeine. This may be explained by the consistently poorer
bioavailability of dihydrocodeine (20 per cent), which probably results
from hepatic presystemic metabolism.(38)
The usual starting dose is 30 mg every 4–6 h (by mouth),
and this may be increased to 60 mg. However, dihydrocodeine appears to
have a narrower therapeutic index than codeine, with a high incidence
of adverse effects at the 60-mg dose. A controlled-release formulation
of dihydrocodeine is available in several countries.
There have been a number of reports of severe toxicity associated with dihydrocodeine in patients with impaired renal function.(39,40)
The mechanism is not clear because of the limited data available on the
pharmacokinetics of this drug, although it seems most likely that the
cause is accumulation of active glucuronide metabolites, as occurs with
morphine.
There is confusion about the relative analgesic potency
of dihydrocodeine. It seems reasonable to assume that oral
dihydrocodeine is roughly equipotent to oral codeine, and to use a
similar conversion ratio when changing to morphine.
Dextropropoxyphene
Propoxyphene is a synthetic derivative of methadone, and
its dextrorotatory stereoisomer dextropropoxyphene is responsible for
its analgesic activity. Dextropropoxyphene is a µ agonist with low
receptor affinity similar to that of codeine. It is readily absorbed
from the gastrointestinal tract with peak serum levels about 2 h after
administration. The mean elimination half-life is about 12 h, with
steady state levels being reached after 3–4 days of regular
administration every 6–8 h. The half-life may be very long (over 50 h)
in elderly patients.(41)
Dextropropoxyphene undergoes extensive first-pass
metabolism. Its principal metabolite is norpropoxyphene, which is
active but penetrates the brain to a much lesser extent and has much
weaker opioid effects. Norpropoxyphene has a longer half-life (about 23
h) than dextropropoxyphene itself and
accumulates in plasma.(42) Norpropoxyphene accumulation is associated with excitatory effects, including tremulousness and seizures.
P.322
accumulates in plasma.(42) Norpropoxyphene accumulation is associated with excitatory effects, including tremulousness and seizures.
The analgesic efficacy and relative potency of
dextropropoxyphene have been questioned. This is in part because
single-dose studies comparing aspirin, paracetamol, and non-steroidal
anti-inflammatory drugs (NSAIDs), including ibuprofen 400 mg, mefenamic
acid 250 mg, and fenoprofen 50 mg, have shown dextropropoxyphene to be
a less effective analgesic.(43) A more recent
systematic review found that paracetamol alone is as effective as the
combination of paracetamol with dextropropoxyphene,(44)
though the studies included were again all single-dose studies.
Single-dose studies may be misleading, as is the case with single-dose
studies of oral morphine.
The extensive first-pass metabolism of
dextropropoxyphene is dose dependent such that the systemic
availability of the drug increases with increasing oral doses.(45)
Thus, with regular administration, there is enhanced bioavailability
and some degree of accumulation because of the long elimination
half-lives of the parent drug and its main metabolite. Both
dextropropoxyphene and norpropoxyphene reach plasma concentrations in
the steady state which are five to seven times greater than those found
after the first dose. There is therefore a pharmacokinetic basis for
believing that repeated doses of dextropropoxyphene are likely to be
more effective than single doses. The usual starting dose of morphine
for patients receiving dextropropoxyphene–paracetamol combinations
every 4–6 h (representing 260–390 mg of dextropropoxyphene daily) is 60
mg per day.
Toxicity of dextropropoxyphene
For a long time a combination of dextropropoxyphene and
paracetamol was the most commonly prescribed analgesic in the United
Kingdom and some Scandinavian countries, but it received much adverse
publicity because of its lethal effects in overdose and fears about its
addiction potential. Part of this concern was stimulated by its very
widespread use. In addition to usual opioid adverse effects,
propoxyphene may rarely induce a hepatotoxic reaction,(46) cardiac conduction disorder,(47)
and potentially dangerous drug interactions have been reported when
propoxyphene has been administered along with carbamazepine,(48) warfarin,(49) or alcohol.(50)
At present, however, there is insufficient evidence to conclude that
dextropropoxyphene is inherently more toxic than codeine or other
opioids of similar efficacy, nor is there evidence that one is more
effective than another.
Oxycodone
Oxycodone is a semi-synthetic congener of morphine,
which has been on the market for 80 years but until recently was only
available in formulations which effectively circumscribed its use. In
the United States, it has been prescribed in low-dose combination
products with a non-opioid for oral administration (usually 5 mg of
oxycodone with either aspirin or paracetamol) and has traditionally
been used as a step 2 analgesic. In the United Kingdom and some other
countries only a rectal suppository and no oral formulations have been
available, so that it has been used only for patients unable to take
oral medication.(51) Oxycodone has been more
widely used by mouth as a first line step 3 opioid in Scandinavia where
it has also been widely used as a post-operative opioid.(52)
Recently, oxycodone has been produced as a single agent in new oral
formulations, both normal release and sustained release, which has
substantially improved the convenience of administration. In many
countries sustained release oxycodone is available in 5, 10, 20, 40,
and 80 mg formulations with a corresponding range of normal release
formulations. Oxycodone is increasingly used as a step 3 opioid,(53,54 and 55)
though the low-dose formulations allow its use at step 2 also.
Oxycodone probably provides the best example of the overlap in efficacy
between opioids at steps 2 and 3.
Tramadol
Tramadol is a centrally acting analgesic which possesses
opioid agonist properties and may also activate monoaminergic spinal
inhibition of pain.(56) It has modest affinity
with µ opioid receptors, with weak affinity to δ and κ receptors, and
its analgesic effect is reversed by naloxone. Unlike other opioids, it
also inhibits the uptake of noradrenaline and serotonin, and in an
animal model systemically administered yohimbine or ritanserin blocks
tramadol-induced analgesia,(56) suggesting that this effect contributes significantly to the drug's analgesic action.
Tramadol can be administered orally, rectally,
intravenously, subcutaneously, or intramuscularly. In many countries,
it is available in both normal- and sustained-release formulations.
Parenterally, 50–150 mg of tramadol is equianalgesic to 5–15 mg
morphine.(57) There are insufficient data for a
reliable assessment of its oral to parenteral relative analgesic
potency and estimates range from 1 : 4(58) to 1 : 10.(59)
Recent studies have demonstrated the efficacy of oral tramadol in the management of chronic cancer pain of moderate severity.(58,59 and 60) Few patients with severe pain are adequately managed by tramadol.(59,61)
Tramadol has a similar side-effect profile to morphine, but may cause
less constipation and respiratory depression at equianalgesic doses.
Opioids in combination with non-opioids
The second step of the analgesic ladder
By convention, formulations combining aspirin or
paracetamol with a low dose of codeine, oxycodone, or propoxyphene have
been recommended for pain of moderate intensity (step 2 of the
analgesic ladder). This recommendation was pragmatic rather than
evidence based. It reflected the concern that in many parts of the
world it would be unacceptable to use morphine or other potent opioids
for moderate pain.
Overall, these combination preparations have frequently
proved to be relatively ineffective because the dose of opioid was too
low (e.g. codeine 8 or 16 mg). Indeed, in the validation studies of the
WHO ladder few patients using these agents maintained adequate relief
for more than a few weeks.(17) Additionally,
these formulations all have a short duration of effect and require
patients to use repeated doses every 3–4 h to achieve continuous
analgesia in the setting of chronic pain.
The most frequently employed step 2 analgesics in cancer
pain are combination preparations containing 300–500 mg paracetamol
with 30 mg codeine, 32.5 mg dextropropoxyphene, or 5 mg oxycodone. The
combination of dextropropoxyphene with paracetamol (coproxamol in the
United Kingdom) has theoretical disadvantages of pharmacokinetic
incompatibility (dextropropoxyphene has a much longer elimination
half-life than paracetamol) and accumulation of dextropropoxyphene and
its active metabolite norpropoxyphene. However, neither problem appears
to have any clinical consequence in practice and this combination
remains widely used. Codeine and paracetamol are pharmacokinetically
more compatible, and at present either combination would be an
appropriate choice.
Recent studies comparing single doses of
opioid/non-opioid combinations with various NSAIDs in post-operative
pain have shown advantages for the latter in terms of greater efficacy
and less adverse effects.(62,63 and 64)
Chronic use of NSAIDs may negate any advantage in terms of unwanted
effects, although at present there are no comparative data for chronic
cancer pain. NSAIDs are increasingly employed as step 2 analgesics.
Given the limitations of the conventional approach many
clinicians now use a variety of single agent opioid agonists, some
previously designated as ‘step 3’ opioids, in an appropriate dose, for
moderate pain. Over recent years, sustained-release formulations of
oxycodone, tramadol and morphine in dose formulations appropriate for
pain of moderate severity have become widely available and are now
often used in this setting. This practice is supported by evidence of
efficacy.(54,60,65)
The partial agonist opioid buprenorphine also may be
used in this setting since it has recently become available in a
transdermal formulation. Preliminary experience has been reported in
the management of moderate cancer pain.(66) A
low-dose formulation of transdermal fentanyl is also under development
and is designed for use in patients who may be opioid naive. There are
potential dangers in the earlier use of the most potent opioids,
particularly when administered in long acting formulations and
more clinical trial data are required to clarify some of the issues surrounding these trends in opioid prescribing.
P.323
more clinical trial data are required to clarify some of the issues surrounding these trends in opioid prescribing.
Opioids for moderate to severe pain
Morphine-like agonists
The morphine-like agonist drugs (Table 2)
are widely used to manage cancer pain. Although they may differ from
morphine in quantitative characteristics they qualitatively mimic the
pharmacological profile of morphine, including both desirable and
undesirable effects. Controversy has developed over the choice of an
opioid drug, in part because of the dearth of well-controlled studies
comparing the efficacy and side-effects of these drugs during chronic
administration, and in part because of the large amount of survey data
and anecdotal reports supporting one drug over another.
Clinicians in most countries have some choice when
selecting which opioid drug(s) to prescribe, but the options can vary
from country to country.
Morphine is still considered to be the opioid drug of choice by many practitioners(30)
and has occupied this place throughout recorded history. However, some
of the other potent µ receptor agonists are gaining popularity for a
variety of reasons. While greater choice is very important in analgesic
therapy, it should not be forgotten that morphine is an excellent
analgesic and maintains a key position and reference point for all
opioids in our therapeutic armamentarium. Similarly, while there is
theoretical evidence (on the basis of receptor binding profiles) that
opioid combinations may give optimum analgesia with a more favourable
side-effect profile, there is as yet no good clinical evidence to
support such treatment strategies which are likely to be appropriate
only in rare situations.
Morphine
Morphine is a potent µ-agonist drug that was first
introduced into clinical use almost 200 years ago. It is the main
naturally occurring alkaloid of opium derived from the poppy Papaver somniferum
and is available for therapeutic use as the sulphate, hydrochloride,
and tartrate. Recent evidence suggests that biosynthetic pathways for
morphine exist in animal and human tissues such as liver, blood, and
brain.(67) Its chemical structure is shown in Fig. 4.
The WHO has placed oral morphine on the Essential Drug List, and
preparations are available for oral, rectal, parenteral, and
intraspinal administration.
Bioavailability
Morphine is available in four oral formulations: an
elixir, a normal-release tablet, a modified-release tablet or capsule
(of which there are now several preparations using different
sustained-release mechanisms), and sustained-release suspensions.
Absorption of morphine after oral administration occurs predominantly
in the alkaline medium of the upper small bowel (morphine is a weak
base) and is more or less complete. After oral administration,
extensive presystemic elimination of the drug occurs predominantly in
the liver. In healthy volunteers and cancer patients, the average
bioavailability for oral morphine is 20–30 per cent.(68,69 and 70)
Like all other pharmacokinetic parameters, bioavailability demonstrates
marked inter-individual variability. In patients with normal renal
function the plasma half-life (2–3 h) is somewhat shorter than the
duration of analgesia (4–6 h). The pharmacokinetics remain linear with
repetitive administration, and there does not appear to be
autoinduction of biotransformation even following large chronic doses.(71)
Morphine is relatively hydrophilic and, when
administered epidurally or intrathecally, it is not rapidly absorbed
into the systemic circulation. This results in a long half-life in
cerebrospinal fluid (90–120 min) and extensive rostral redistribution.(72)
Metabolism
About 90 per cent of morphine is converted into metabolites (Fig. 5),
principally the glucuronide conjugates morphine-3-glucuronide (M3G) and
morphine-6-glucuronide (M6G); minor metabolites include codeine,
normorphine, and morphine ethereal sulfate. The liver appears to be the
predominant site of metabolism in humans, although in animal models
extrahepatic metabolism has been demonstrated in the small bowel and
the proximal renal tubule of rodents. These sites may become important
where liver function is impaired. M3G is the major metabolite and in
recent years there has been some controversy about its possible role as
an opioid antagonist or in mediating some of the adverse effects of
morphine.
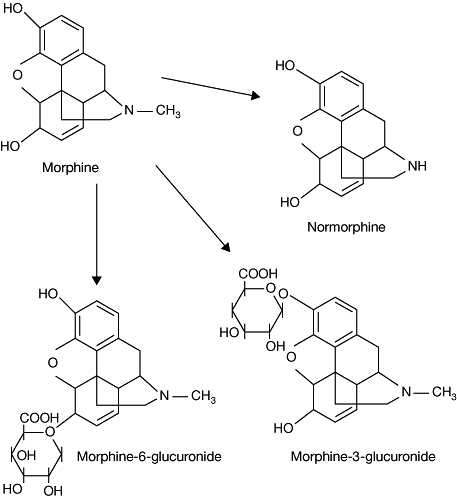 |
Fig. 5 The metabolites of morphine. |
Morphine-6-glucuronide
M6G binds to opioid receptors(73) and produces potent opioid effects in animals(73,74 and 75) and humans.(73,76,77 and 78) M6G excretion by the kidney is directly related to creatinine clearance;(79)
its elimination half-life is 2–3 h in patients with normal renal
function (similar to that of morphine) but becomes progressively longer
with deteriorating function, resulting in significant accumulation.(79) In patients with impaired renal function, M6G may accumulate in blood and cerebrospinal fluid,(80) and high concentrations of this metabolite have been associated with toxicity.(76,81)
Although further studies are needed to clarify the clinical importance
of M6G and other metabolites, the data available are sufficient to
recommend caution when administering morphine to patients with renal
impairment. Patients who are receiving regular morphine and develop
acute renal failure in a previously stable situation (e.g. a rapidly
developing obstructive uropathy in a patient with pelvic malignancy)
may develop a sudden onset of signs and symptoms of opioid toxicity,
necessitating temporary withdrawal of the morphine and subsequent dose
reduction, and/or less frequent administration.
M6G is thought to be a potent analgesic and studies in
acute post-operative pain are currently ongoing. It is not yet clear
whether M6G will have fewer side-effects than morphine, though it has
been suggested that M6G causes less respiratory depression.(82,83 and 84)
Morphine-3-glucuronide
For many years, it has been assumed that M3G is inert as is the case with most glucuronide metabolites.(85) Recent behavioural studies in rodents, however, suggested that M3G produces a functional antagonism of the
analgesic effects of morphine and its active metabolite M6G.(86,87) There is also some evidence in animal models that M3G may be responsible for the central nervous system excitatory adverse effects seen with morphine, such as myoclonus.(88,89)
P.324
analgesic effects of morphine and its active metabolite M6G.(86,87) There is also some evidence in animal models that M3G may be responsible for the central nervous system excitatory adverse effects seen with morphine, such as myoclonus.(88,89)
It is now clear that M3G does not bind to opioid
receptors. Data from electrophysiological animal models indicate no
evidence of an antagonistic effect of M3G(90) and recent studies in human volunteers indicate that M3G appears to be devoid of significant activity.(83,91)
In particular, there is no evidence of functional antagonism of
morphine or M6G in humans and overall it seems that M3G plays no
significant role in the pharmacodynamics of morphine.
Oral to parenteral relative potency
Single-dose studies of morphine in post-operative cancer patients demonstrated an oral-to-intramuscular potency ratio of 1 : 6.(92)
However, empirical clinical practice using chronically administered
oral morphine in cancer patients has generated a different ratio of 1 :
3 or 1 : 2.(93,94) The
reason for the discrepancy between relative potency estimates derived
from single-dose versus chronic dosing studies is probably associated
with both methodology(95) and the pharmacokinetics and pharmacodynamics of M6G.(93)
It is possible that M6G accumulation relative to morphine may be
greater with oral than with parenteral administration; this would lead
to an increase in the relative potency of the orally administered drug
when given on a chronic basis.
The important principle for clinical practice is that
there is a difference in relative analgesic potency when the route of
administration is changed, and that adjustment of dose is necessary in
order to achieve an equivalent effect and to avoid either underdosing
or toxicity. The usual practice when converting from oral morphine to
subcutaneous morphine (or diamorphine) is to divide the oral dose by
two or three.(30)
Parenteral morphine
The inorganic salts of morphine (morphine sulphate and
morphine hydrochloride) have limited solubility. Standard formulations
are available up to 20 mg/ml, and morphine can be constituted from
lyophilized power up to 50 mg/ml. Morphine tartrate is substantially
more soluble and, in some countries, is formulated in a concentration
of 80 mg/ml.
Sustained-release morphine preparations
The development of modified-release morphine
preparations has had a major impact on clinical practice. These
preparations, which are usually administered on a 12-h schedule,
provide a much more convenient means of administering oral morphine.(96)
Several preparations are available worldwide with a range of dose
formulations (10, 15, 30, 60, 100, and 200 mg depending on the
country), allowing considerable flexibility in their use. Some
preparations allow once-daily administration and sustained-release
suspensions are also available.(97)
In contrast with morphine solution or normal-release
tablets, where peak plasma concentrations are achieved within the first
hour followed by a rapid decline and an elimination half-life of 2–4 h,
sustained-release morphine typically achieves peak plasma
concentrations 3–6 h after administration, the peak is attenuated, and
plasma concentrations are sustained over a 12- or 24-h period.(98,99 and 100)
The type and incidence of adverse effects with sustained-release
morphine and normal-release oral morphine appear to be similar with the
currently available formulations.
Although some clinicians advocate the use of
sustained-release morphine when initiating morphine therapy in cancer
patients, a normal-release preparation is generally recommended in the
dose titration period.(30) Initial dose
titration using sustained-release morphine is difficult because of the
delay in achieving peak plasma concentrations, the attenuation of peak
concentrations, and the long duration of action. In this situation,
dose finding is performed more efficiently with a short-acting morphine
preparation. Once the effective dose is identified using a
normal-release formulation, this may be changed to a sustained-release
preparation using a milligram-to-milligram conversion. For the same
reasons, sustained-release morphine is not appropriate for the
treatment of acute pain or ‘breakthrough’ pain. A normal-release
morphine preparation should be provided to patients stabilized on
sustained-release morphine to be used ‘as required’ for breakthrough
pain.
Diamorphine (heroin)
Diamorphine (diacetylmorphine) is a semi-synthetic
analogue of morphine and has a long tradition of use for cancer pain in
the United Kingdom. It is only available for legal medicinal use in the
United Kingdom and Canada.
Following oral administration of diamorphine, only
morphine can be measured in the patient's blood. The use of oral
diamorphine is an inefficient way of delivering morphine to the
systemic circulation. There is no good basis to believe that there is
any difference between these two drugs when given by mouth. Sublingual
administration of diamorphine has been advocated by some but, as
discussed below, this route is not appropriate for either morphine or
diamorphine because of poor absorption.
It has been thought that diamorphine does not itself
bind to the µ opioid receptor but must be biotransformed to
6-acetylmorphine and morphine to produce its analgesic effect.(101)
However, recent studies with mor-knockout mice seem to indicate that it
does not produce its effects through µ receptor binding and may have
effects at other receptors.(102) This may explain some of the pharmacodynamic differences between morphine and diamorphine when given parenterally.
Since diamorphine is more soluble and lipophilic than
morphine, it does have some advantages for parenteral administration.
When administered by subcutaneous or intramuscular injection,
diamorphine is approximately twice as potent as morphine. There are
also differences between diamorphine and morphine administered by
intravenous injection: diamorphine has a marginally quicker onset of
action, produces greater sedation, and possibly less vomiting.(103)
This may be explained by different receptor binding. The greater
solubility of diamorphine (shared also with hydromorphone and morphine
tartrate) is of particular advantage for patients who require large
doses of subcutaneous opioids.
Methadone
Methadone is a synthetic opioid with an
oral-to-parenteral potency ratio of 1 : 2 and an oral bioavailability
greater than 85 per cent. In single-dose studies, methadone is only
marginally more potent than morphine; however, with repeated
administration it is several times more potent. Methadone has a very
long plasma half-life, averaging approximately 24 h (with a range from
12 to over 150 h).(104,105)
Whereas most patients can be well controlled on 8–12-h dosing, some
patients require dosing at a 4–8-h interval to maintain analgesic
effects.(106) Methadone may be a useful
alternative to morphine, but its safe administration requires knowledge
of its pharmacology and experience of its use.
After treatment is initiated or the dose is increased,
plasma concentration rises over a prolonged period, and this may be
associated with a delayed onset of side-effects. Consequently, patients
must be followed closely until there is reasonable certainty that a
steady state plasma concentration has been approached (approximately 1
week). Serious adverse effects can be avoided if the initial period of
dosing is accomplished with ‘as needed’ administration.(107)
When steady state has been achieved, scheduled dose frequency should be
determined by the duration of analgesia following each dose.(108)
Oral and parenteral preparations of methadone are available. Subcutaneous infusion is possible(109) but caution is required since local skin toxicity may be a problem.(110)
The equianalgesic dose ratio of morphine to methadone
has been a matter of confusion and controversy. Recent data from
crossover studies with morphine and methadone and hydromorphone and
methadone indicate that methadone is much more potent than previously
described in literature, and that the ratio correlates with the total
opioid dose administered before switching to methadone.(111) Among patients receiving low doses of morphine, the ratio is 4 : 1. In contrast, for patients receiving more
than 300 mg of oral morphine (or parenteral equivalent) the ratio is approximately 10 : 1 or 12 : 1.(111)
P.325
than 300 mg of oral morphine (or parenteral equivalent) the ratio is approximately 10 : 1 or 12 : 1.(111)
Pethidine (meperidine)
Pethidine is a synthetic opioid with agonist effects
similar to those of morphine but a profile of potential adverse effects
that limits its utility as an analgesic for chronic cancer pain.
Intramuscular pethidine 75 mg is equivalent to 10 mg of intramuscular
morphine. Pethidine has an oral bioavailability of 40–60 per cent, and
its oral-to-parenteral potency ratio is 1 : 4. It is more lipophilic
than morphine, and produces a faster onset and shorter duration of
analgesia of 2–3 h.
Pethidine is N-demethylated
to norpethidine, which is an active metabolite that is twice as potent
as a convulsant and half as potent as an analgesic compared with its
parent compound. Accumulation of norpethidine after repetitive dosing
of pethidine can result in central nervous system excitability
characterized by subtle mood effects, tremors, multifocal myoclonus,
and occasionally, seizures.(112,113)
Naloxone does not reverse pethidine-induced seizures, and it is
possible that its administration to patients receiving pethidine
chronically could precipitate seizures by blocking the depressant
action of pethidine and allowing the convulsant activity of
norpethidine to become manifest.(114) If
naloxone is necessary in this situation, it should be diluted and
slowly titrated while appropriate seizure precautions are taken.
Selective toxicity of pethidine can also occur following administration
to patients receiving monoamine oxidase inhibitors. This combination
may produce a syndrome characterized by hyperpyrexia, muscle rigidity,
and seizures which may occasionally be fatal.(115) The pathophysiology of this syndrome is related to excess availability of serotonin at the 5HT1A-receptor in the central nervous system.
Although accumulation of norpethidine is most likely to
affect patients with overt renal disease, toxicity is sometimes
observed in patients with normal renal function. These potential
adverse effects contraindicate pethidine for the management of chronic
cancer pain.
Given the availability of alternative drugs that lack these toxicities, its use in acute pain management is also not recommended.(117)
P.326
Given the availability of alternative drugs that lack these toxicities, its use in acute pain management is also not recommended.(117)
Hydromorphone
Hydromorphone is another morphine congener. It is five
times more potent than morphine and can be administered by the oral,
rectal, parenteral, and intraspinal routes. Its oral bioavailability
varies from 35 to 80 per cent, and its oral-to-parenteral potency ratio
is 1 : 5.(118) Its half-life is 1.5–3 h and it
has a short duration of action. Although it is largely excreted
unchanged by the kidney, it is partially metabolized in the liver to a
3-glucuronide, which is excreted by the kidneys.(118,119)
Its solubility, the availability of a high-concentration
preparation (10 mg/ml), and high bioavailability by the subcutaneous
route (78 per cent) make it particularly suitable for subcutaneous
infusion.(120) In the United States, it is
routinely available in oral, rectal, and injectable formulations, and a
sustained-release oral formulation.(121) For
patients who require very high opioid doses via the subcutaneous route,
hydromorphone can be constituted in concentrations of up to 50 mg/ml
from lyophilized powder. It has also been administered via the epidural
and intrathecal routes to manage acute and chronic pain. Hydromorphone
is hydrophilic and, when administered via the epidural route, its
pharmacokinetic profile, including its long half-life and extensive
rostral distribution in cerebrospinal fluid, is similar to that of
morphine.(122)
The equianalgesic ratio of parenteral morphine to
hydromorphone has become a matter of controversy; recent data suggests
that for chronic dosing it is less than the traditionally quoted ratio
of 1 : 7 and that it is probably closer to 1 : 4.(123,124)
Levorphanol
Levorphanol is a morphine congener with a long half-life (12–16 h).(125) It is five times more potent than morphine and has an oral-to-parenteral potency ratio of 1 : 2.(126)
Like methadone, the discrepancy between plasma half-life (12–16 h) and
duration of analgesia (4–6 h) may predispose to drug accumulation
following the initiation of therapy or dose escalation. Although dose
titration needs to be done carefully in the opioid-naive patient,
problems with drug accumulation appear to be less than those produced
by methadone.
In the United States, levorphanol is generally used as a
second-line agent in patients with chronic pain who cannot tolerate
morphine. The possibility that this drug may be particularly useful in
morphine-tolerant patients has been proposed on the basis of its
affinity for receptors κ3 and δ that are presumably not involved in
morphine analgesia.(127) It is no longer available in the United Kingdom or Canada.
Oxycodone
As previously described, oxycodone is a synthetic
morphine congener that has a high oral bioavailability (60–90 per cent)
and an analgesic potency 30–50 per cent greater than morphine.(128,129)
Since the development of sustained release formulations in doses
suitable for severe pain, it is now widely used for this indication.
The sustained release formulation is available in a wide range of dose
formulations (5, 10, 20, 40, and 80 mg)(55) and
has a duration of action of 8–12 h. The sustained-release formulation
achieves effective therapeutic levels within an hour(130) and appears to be suitable for dose titration.(131)
Oxycodone pectinate is available in the United Kingdom as a 30-mg
rectal suppository which has a delayed absorption and prolonged
duration of effect.(51)
There has been confusion about the relative efficacy of
oxycodone. Until recently, it has been viewed primarily as a ‘step 2’
opioid because it has for long been available in low dose in
combination products with non-opioid analgesics. It seems clear that
the relative potency of oxycodone has been underestimated in early
clinical studies in which it appeared to be less potent than morphine.
As indicated above more recent studies indicate that it is more potent,
in a ratio of about 1.5 : 1.(128)
There remains uncertainty also about the role of its
active metabolite oxymorphone in mediating the effects of oxycodone.
However, current evidence suggests that the metabolites of oxycodone
including oxymorphone do not contribute significantly to its
pharmacological effects.(11)
Oxymorphone
Oxymorphone is a lipophilic congener of morphine. It is
currently most widely used in suppository form, infrequently used
parenterally on a chronic basis, and is not available orally. The
injectable formulation is 10 times more potent than morphine.(132)
A rectal formulation that is approximately equipotent with parenteral
morphine is also available in the United States. The plasma half-life
of oxymorphone is 1.2–2 h, and its duration of action is 3–5 h. It is
less likely to produce histamine release than morphine,(133) and may be particularly useful for patients who develop itch in response to other opioids.(134) Oxymorphone is currently not available in the United Kingdom.
Fentanyl
Fentanyl is a semisynthetic opioid and is a highly selective µ agonist(135)
that is about 80 times as potent as parenteral morphine in the
non-tolerant acute pain patient. It is also extremely lipophilic and is
extensively taken up into fatty tissue.(136)
Its elimination half-life ranges from 3 to 12 h and is influenced by
the duration of prior administration and the extent of fat
sequestration. Fentanyl has been used mainly as an intravenous
anaesthetic agent and continues to be used parenterally as a
pre-medication for painful procedures and in continuous infusions. When
used intravenously, fentanyl has a very short duration of action of
0.5–1 h. This is related to the rapid redistribution of the drug into
body tissues rather than to hepatic and renal elimination.(137)
The development of a transdermal system and an oral transmucosal
formulation has broadened the clinical utility of fentanyl for the
management of cancer pain.
Transdermal fentanyl
The low molecular weight and high lipid solubility of
fentanyl facilitate absorption through the skin and a transdermal
formulation that delivers 25, 50, 75, or 100 µg/h is widely available.(138,139 and 140)
The transdermal system consists of a drug reservoir that is separated
from the skin by a copolymer membrane that controls the rate of drug
delivery to the skin surface. The drug is released at a nearly constant
amount per unit time along a concentration gradient from the patch to
the skin. After application of the transdermal system, serum fentanyl
concentration increases gradually, usually levelling off after 12–24 h,
and then remaining stable for a time before declining slowly. When the
patch is removed, serum concentration falls 50 per cent in
approximately 17 h (range 13–22 h).(141) The
slow onset of effect after application and an equally slow decline in
effect after removal are consistent with the development of a
subcutaneous depot of drug that maintains the plasma concentration.
There is significant interindividual variability in fentanyl
bioavailability by this route and dose titration is necessary.(141)
The dosing interval for each system is usually 72 h, but
interindividual pharmacokinetic variability is large and some patients
require a dosing interval of 48 h.(142)
Familiarity with the kinetics of the transdermal system
is essential for optimal use. Since there is a delay of 8–12 h in
achieving effective analgesia after initial application of the patch,
it is necessary to provide alternative analgesia for this initial
period. It is prudent to apply the patch in the early hours of the day
so that the patient can be observed as blood levels rise over the
ensuing 12 h to minimize the risk of overdosing during sleep.
Significant concentrations of fentanyl can remain in the plasma for up
to 24 h after removal of the patch because of delayed release from
tissue and subcutaneous depots. Neither age nor patch location appears
to affect fentanyl absorption from the transdermal system.(140)
There is a potential for temperature-dependent increases in fentanyl
release from the system associated with increased skin permeability in
patients with fever, who should be monitored for opioid side-effects.
Patients should also avoid exposing the patch to direct external heat.
Empirically, the indications for the transdermal route
include intolerance of oral medication, poor compliance with oral
medication, and occasionally the desire to provide a trial of fentanyl
to patients who have reacted unfavourably to other opioids. However,
there are a number of limitations. The delay in onset of analgesia and
in the establishment of steady state blood levels require the liberal
use of an alternative short-acting opioid (usually morphine) for
breakthrough pain during the early treatment period. Because of its
3-day duration of action, transdermal fentanyl is generally unsuitable
for patients with unstable pain, and if a patient's pain goes out of
control management may be complicated because of the delay in
re-establishing steady state. If dose reductions are required or
discontinuation is indicated, the continuing absorption following patch
removal must be taken into account. Poor patch adhesion may be a
problem in some patients. Set against these considerations are the
advantages in terms of convenience and compliance and there is high
patient acceptability of this mode of administration. Additionally
there are experimental and clinical data to suggest that transdermal
fentanyl is associated with less constipation than morphine.(143,144)
Empirical observations suggest that a 100-µg/h fentanyl
patch is approximately equianalgesic to 2–4 mg/h of intravenous
morphine (or equivalent). The relative potency ratio that is applicable
when converting patients from oral morphine to transdermal fentanyl has
been the subject of some controversy, but the dosing recommendations of
the manufacturer seem about right. The patch should be placed in an
area where skin movement is limited, such as the upper anterior chest
wall or either side of the midline on the back, preferably the lower
back. Studies have shown that all areas of skin absorb the drug at
roughly the same rate.(140) Since the adhesive
strips on these patches are less than optimal, securing the patch with
non-irritant tape is often necessary.
Transdermal fentanyl is best reserved for patients whose opioid requirements are stable(30) and in general it is likely to be a second-line choice. However, for suitable patients it works well and they like it.(145)
Oral transmucosal fentanyl citrate (OTFC)
An oral transmucosal formulation of fentanyl (which
incorporates the drug in a hardened lozenge on a stick) that is
absorbed across the buccal mucosa, has recently been introduced in many
countries for the management of breakthrough pain. The lozenge is
rubbed gently against the inside of the cheek until it has dissolved.
The formulation is rapidly absorbed and achieves blood levels and time
to peak effect that are comparable to parenterally administered
fentanyl. Indeed, the time to onset of analgesia is 5–10 min(146,147,148 and 149)
and studies in cancer patients suggest that it can provide rapid and
very effective relief of breakthrough pain. Formulations incorporating
200, 400, 600, 800, and 1600 µg are available. The most common adverse
effects associated with this formulation are somnolence, nausea, and
dizziness. One interesting observation which has emerged from the
clinical trials and clinical use is that the successful dose of OTFC
cannot be predicted and is not directly related to the daily dose of
regular opioids being received for background pain. This raises some
questions about the current management of breakthrough pain with
conventional formulations of oral or parenteral opioids (see Chapter 8.2.11). The use of OTFC is still relatively limited but initial experience has been good.
Other drugs
Phenazocine
Phenazocine is a synthetic opioid structurally related
to morphine with strong binding to the σ receptor. One 5-mg tablet is
equivalent to 25 mg of oral morphine, which means that there is less
flexibility in its use. Phenazocine may be given sublingually, although
administration by this route is usually avoided because of its bitter
taste and variable absorption. In the United Kingdom, it was often used
for patients who are unable to tolerate oral morphine, but it is now
largely replaced by the more recently available alternative opioids
such as oxycodone, fentanyl, and hydromorphone.
Dextromoramide
Dextromoramide is a µ agonist and is approximately twice
as potent as morphine when taken by mouth. Few data on its
pharmacokinetics are available because of difficulties in accurately
assaying the drug, but in clinical practice in cancer pain it has a
rapid onset of action but a shorter duration than morphine. Tolerance
to dextromoramide seems to develop rapidly in humans and, although the
duration of analgesia may initially be 2–4 h, with repeated
administration this may be reduced to only 1 or 2 h. For this reason,
it is unsuitable for maintenance treatment in chronic cancer pain,
although it has been used successfully as a short-acting strong
analgesic for breakthrough pain in some patients. It does not have any
particular advantages over morphine used in this way, and in general
the use of dextromoramide in chronic cancer pain is not recommended.
Dipipanone
Dipipanone is a diphenylpropylamine structurally related
to both dextromoramide and methadone. As an analgesic, it is
approximately half as potent as morphine, and in the United Kingdom it
is only available in a combination tablet containing 10 mg dipipanone
and 30 mg cyclizine. For many patients this results in excessive
sedative and anticholinergic side-effects related to cyclizine when
adequate analgesic doses are given, and thus it has only limited
application in the management of chronic cancer pain.(150)
Agonist–antagonist opioid analgesics
The agonist–antagonist opioid analgesics are a
heterogeneous group of drugs with moderate to strong analgesic
activity, comparable with that of the agonist opioids such as codeine
and morphine. The group includes drugs which act as an agonist or
partial agonist at one receptor and as an antagonist at another
(pentazocine, dezocine, butorphanol, nalbuphine)—‘the mixed
agonist–antagonists’—and drugs acting as a partial agonist at a single receptor (buprenorphine). These two groups of drugs can be also classified as nalorphine- or morphine-like. Meptazinol fits neither classification and occupies a separate category. The place of this group of drugs in chronic cancer pain has been limited.(151) However, the recent development of a transdermal formulation of buprenorphine may allow its more widespread use in both chronic cancer and non-cancer pain.
P.327
agonist–antagonists’—and drugs acting as a partial agonist at a single receptor (buprenorphine). These two groups of drugs can be also classified as nalorphine- or morphine-like. Meptazinol fits neither classification and occupies a separate category. The place of this group of drugs in chronic cancer pain has been limited.(151) However, the recent development of a transdermal formulation of buprenorphine may allow its more widespread use in both chronic cancer and non-cancer pain.
Mixed agonist–antagonist analgesics
The agonist–antagonists produce analgesia in the
opioid-naive patient but may precipitate withdrawal in patients who are
physically dependent on morphine-like drugs. Therefore, when used for
chronic pain, they should be tried before repeated administration of a
morphine-like agonist drug.
Pentazocine, butorphanol, and nalbuphine are µ
antagonists and κ agonists or partial agonists. All three drugs are
strong analgesics when given by injection: pentazocine is one-sixth to
one-third as potent as morphine, nalbuphine is roughly equipotent with
morphine, and butorphanol is 3.5–7 times as potent. The duration of
analgesia is similar to that of morphine (3–4 h). Oral pentazocine is
closer in analgesic efficacy to aspirin and paracetamol than the weak
opioid analgesics, such as codeine. Neither nalbuphine nor butorphanol
is available as an oral formulation, and butorphanol is no longer
available in any form in the United Kingdom.
At usual therapeutic doses, nalbuphine and butorphanol
have respiratory depressant effects equivalent to that of morphine
(although the duration of such effects may be longer with butorphanol).
Unlike morphine, there appears to be a ceiling to both the respiratory
depression and the analgesic action.
All three drugs have a lower abuse potential than the
agonist opioid analgesics such as morphine. However, all have been
subject to abuse and misuse, and pentazocine (but not the others) is
subject to controlled drug restrictions. In North America, the oral
preparation of pentazocine is marketed in combination with naloxone
(but is available without naloxone elsewhere).
Meptazinol is a synthetic hexahydroazepine derivative
with opioid agonist and antagonist properties, but is unlike either the
nalorphine-type agonist–antagonists or buprenorphine. Meptazinol has
central cholinergic properties which may account at least in part for
its analgesic effects. Receptor binding studies show it to be a
specific µ1 agonist. Meptazinol is one-tenth as potent as morphine by
intramuscular injection and has a duration of action of about 4 h. Some
studies have shown adverse effects to be more frequent than with
morphine, although respiratory depression and constipation appear to be
less.
In therapeutic doses, the mixed agonists–antagonists may
produce certain self-limiting psychotomimetic effects in some patients;
pentazocine is the most common drug associated with these effects.
These drugs play a very limited role in the management of chronic
cancer pain because the incidence and severity of the psychotomimetic
effects increase with dose escalation, and nalbuphine and butorphanol
are only available for parenteral use.
A transnasal formulation of butorphanol is now on the
market in the United States, but there is no reported experience of its
use in the management of chronic cancer pain.
Partial agonist analgesics
Buprenorphine (Table 2) is a
semi-synthetic derivative of thebaine and chemically closely related to
the strong agonist etorphine. Buprenorphine is a true partial agonist
at the µ receptor and exhibits a ceiling effect in dose response curves
in various animal models. In some, a bell-shaped curve is seen,
indicating that at doses above a certain level the pharmacological
effect actually decreases with increasing dose.(152)
Buprenorphine has until recently only been available by injection or
for sublingual administration. A dose of 0.4 mg sublingually gives
similar analgesia to 0.2–0.3 mg intramuscularly, with an onset of
analgesia within 30–60 min of administration and a duration of 6–9 h.(153) In contrast, if taken orally, buprenorphine is a poor analgesic due to extensive presystemic elimination.(154)
The long duration of analgesia with buprenorphine may be related to its
affinity for the µ-opioid receptor and an unusually slow dissociation
constant for the drug–receptor complex.
Buprenorphine has been in clinical use for more than 25
years and has been evaluated in a variety of acute pain models. Direct
single dose comparisons with other analgesics such as morphine is
complicated by its long duration of action, but results from a number
of studies in post-operative pain suggest that single doses of 0.3 mg
buprenorphine parenterally or 0.4 mg sublingually give equivalent
analgesia to 10–15 mg intramuscular morphine. A ceiling effect for
analgesia in humans has not been clearly demonstrated.
Buprenorphine produces typical opioid adverse effects.
Overall the available data (which is limited) suggest that the
incidence of common adverse effects compared with morphine is similar.
Naloxone appears to be relatively ineffective in reversing opioid
effects due to buprenorphine.(155)
Co-administration of buprenorphine to patients receiving high doses of
a morphine-like agonist may precipitate withdrawal symptoms.
Buprenorphine was introduced in high-dose sublingual
tablet formulations in 1999 for the management of drug dependence. This
potential use of the drug has long been recognized(156) and it has been suggested that there is less overdose risk compared with other opioids.(157)
Buprenorphine has been recently introduced in a patch
for transdermal administration. The drug is incorporated in a polymer
adhesive matrix (with no liquid reservoir). Three patch sizes are
available delivering 35, 52.5, and 70 µg buprenorphine per hour, and
each lasts for 3 days. Therapeutic plasma concentrations are achieved
within 11–21 h and steady state between the second and third
applications of the patch. At usual clinical doses of 3–4 mg per 24 h
buprenorphine functions as a pure µ agonist.
Transdermal buprenorphine has been licensed for use in
both cancer pain and non-cancer pain but clinical experience with this
formulation is limited.
Principles of opioid administration
The effective clinical use of opioid drugs requires
familiarity with the different drugs available, routes of
administration, dosing guidelines, and potential adverse effects.
Indications
A trial of opioid therapy should be given to all
patients with pain of moderate or greater severity, irrespective of the
underlying pathophysiological mechanism. As discussed in Chapter 8.2.11,
the suggestion that some forms of pain, such as neuropathic pain, are
intrinsically refractory to opioid analgesia has been refuted by
several studies that demonstrate that pain mechanisms do not accurately
predict analgesic outcome from opioid therapy.(158)
Given the variability of response, all opioid trials in the clinical
setting should include dose titration until adequate analgesia occurs
or intolerable adverse effects supervene. This approach will identify
those responders who can gain substantial clinical benefit from opioid
therapy.
Patients whose pain is not easily controlled with an
opioid analgesic because of troublesome adverse effects may benefit
from alternative strategies and these are discussed below.
Drug selection
The factors that influence opioid selection include pain
intensity, pharmacokinetic considerations and available formulations,
previous adverse effects, and the presence of co-existing disease.
Pain intensity
Patients who present with severe pain are usually
treated with a ‘step 3’ opioid (morphine, hydromorphone, oxycodone,
oxymorphone, fentanyl, methadone, or levorphanol). Patients with
moderate pain are conventionally treated with a combination product
containing paracetamol or aspirin plus a conventional step 2 opioid
[codeine, dihydrocodeine, hydrocodone, oxycodone (low dose), and
propoxyphene].
P.328
Pharmacokinetic considerations and type of formulation
Any of the available agonist opioids can be selected for
the opioid-naive patient without major organ failure. Short-half-life
opioids (morphine, hydromorphone, oxycodone, or oxymorphone) are
generally favoured because they are easier to titrate than the
long-half-life drugs, which require a longer period to approach steady
state plasma concentrations. Among the short-half-life opioids, the
range of available formulations often influences specific drug
selection. For ambulatory patients who are able to tolerate oral
opioids, morphine sulphate is generally preferred since it has a short
half-life and is easy to titrate in its normal-release form; it is also
available as sustained-release preparations that allow 12- and 24-h
dosing intervals. The long-half-life opioids methadone and levorphanol
are not usually considered for first-line therapy because they can be
difficult to titrate and present challenging management problems if
delayed toxicity develops as plasma concentrations gradually rise
following dose increments. For the reasons previously described, the
use of pethidine, dextromoramide, and dipipanone for the management of
cancer pain is discouraged.
When the oral route of opioid administration is
contraindicated, the available routes of administration may become an
important consideration in opioid selection. Fentanyl and buprenorphine
are available for administration by the transdermal route. Although
most of the full agonist drugs are well absorbed by subcutaneous
infusion, some (like morphine tartrate, hydromorphone, and diamorphine)
are more suitable by virtue of their high solubility and low
irritability. Methadone and fentanyl may produce significant local
irritation when administered by the subcutaneous route. For cultural
and aesthetic reasons, the subcutaneous route is often preferred to
rectal administration. Subcutaneous infusion may be also preferable in
patients at the end of life because it is less disruptive than using
intermittent analgesic suppositories when nursing a sick patient.
Response to previous trials of opioid therapy
It is always important to review the response to
previous trials of opioid therapy. If the current opioid is well
tolerated, it is usually continued unless difficulties in dose
titration occur or the required dose cannot be administered
conventionally. If dose-limiting side-effects develop, a trial of an
alternative opioid should be considered as discussed in the section on adverse effects.
Co-existing disease
Pharmacokinetic studies of pethidine, pentazocine, and
propoxyphene have revealed that liver disease may decrease the
clearance and increase the bioavailability and half-lives of these
drugs.(159,160) These
changes may result in above-normal plasma concentrations. Mild or
moderate hepatic impairment has only a minor impact on morphine
clearance;(161) however, advanced disease may be associated with reduced elimination.(162)
Patients with renal impairment may accumulate the active
metabolites of propoxyphene (norpropoxyphene), pethidine
(norpethidine), and morphine (M6G). Particular caution is required in
the administration of these drugs to such patients.(76,79,163,164)
Until more data are available, it may be wise to assume that other
opioids with active metabolites may produce similar problems of
toxicity in patients with impaired renal function.
Morphine remains the standard step 3 opioid analgesic
against which others are measured and is the most widely available in a
variety of oral formulations.(30) It has
limitations; the systemic availability of morphine by the oral route is
poor (20–30 per cent) and this contributes to the sometimes
unpredictable onset of action and great interindividual variability in
dose requirements and response. Active metabolites may contribute to
toxicity particularly in patients with renal impairment.(164)
Sometimes the pain does not respond well or completely to morphine,
notably neuropathic pain. However, none of the alternatives to morphine
has so far demonstrated advantages which would make it preferable in
routine use as the first-line oral opioid for cancer pain. Morphine
remains the standard but for reasons of familiarity, availability and
cost rather than proven superiority.
Routes of administration
Opioids should be administered by the least invasive and
safest route capable of providing adequate analgesia. In a survey of
patients with advanced cancer, more than half required two or more
routes of administration prior to death, and almost a quarter required
three or more.(165)
Oral administration
The oral route of opioid administration remains the most
important and appropriate in routine practice. Orally administered
drugs have a slower onset of action, a delayed peak time, and a longer
duration of effect compared with parenterally administered drugs. The
time to peak effect depends on the drug and the nature of the
formulation. For most normal-release oral formulations, peak effect is
typically achieved within 60 min. The oral route of drug administration
is inappropriate for patients who have impaired swallowing or
gastrointestinal obstruction, and for some patients who require a rapid
onset of analgesia. For patients who require very high doses, the
inability to prescribe a manageable oral opioid regimen may be an
indication for the use of a non-oral route.
When given orally, the opioids differ substantially with
respect to their relative analgesic potency compared with parenteral
administration. To some extent, this reflects differences in
presystemic metabolism, that is, the degree to which they are
inactivated as they are absorbed from the gastrointestinal tract and
pass through the liver into the systemic circulation. As indicated in Table 3,
morphine, diamorphine, pethidine, hydromorphone and oxymorphone, have
ratios of oral to parenteral potency ranging from 1 : 3 to 1 : 12.
Methadone, levorphanol, and oxycodone are subject to less presystemic
elimination and also demonstrate a lower oral-to-parenteral potency
ratio of at least 1 : 2. Failure to recognize these differences may
result in a substantial reduction in analgesia when a change from
parenteral to oral administration is attempted without upward titration
of the dose, or toxic effects when changing in the opposite direction.
Rectal administration
The rectal route is a non-invasive alternative to
parenteral routes for patients unable to use oral opioids. Rectal
suppositories containing morphine, hydromorphone, oxymorphone, and
oxycodone are available. The pharmacokinetics and bioavailability of
drugs given rectally may differ from that of oral administration
because of delayed or limited absorption and partial bypassing of
presystemic hepatic metabolism. In practice, however, the potency of
opioids administered rectally is approximately equal to that achieved
by oral dosing.(166) In contrast with morphine, rectal oxycodone appears to have a delayed absorption and prolonged duration of action.
For many patients, the rectal route is not used because
it is more convenient to convert directly to a subcutaneous infusion of
opioid using a portable syringe driver or similar device.
Parenteral administration
Bolus injections
Parenteral routes of administration are considered for
patients who have impaired swallowing or gastrointestinal obstruction,
those who require a rapid onset of analgesia, and those who require
very high doses that cannot be conveniently administered by other
methods. Repeated parenteral bolus injections, which can be delivered
by the intravenous, intramuscular, or subcutaneous routes, may be
complicated by the occurrence of untoward ‘bolus’ effects (toxicity at
peak concentration and/or pain breakthrough at the trough). Intravenous
bolus provides the most rapid onset; the time to peak effect correlates
with the lipid solubility of the opioid, ranging from 2 to 5 min for
methadone and from 10 to 15 min for morphine. Although repetitive
intramuscular injections are commonplace in some countries, they are
painful and offer no pharmacokinetic advantage, and their use is not
recommended.(30,31) Repeated bolus doses, if required, can be accomplished without frequent skin punctures by using an indwelling intravenous
or subcutaneous infusion device. To deliver repeated subcutaneous injections, a 25–27 gauge ‘butterfly’ can be left under the skin for up to a week.(167) The discomfort associated with this technique is partially related to the volume to be injected; it can be minimized by the use of concentrated formulations.
P.329
or subcutaneous infusion device. To deliver repeated subcutaneous injections, a 25–27 gauge ‘butterfly’ can be left under the skin for up to a week.(167) The discomfort associated with this technique is partially related to the volume to be injected; it can be minimized by the use of concentrated formulations.
Continuous infusions
Continuous infusions avoid the problems associated with
the ‘bolus effect’ and can be administered intravenously or
subcutaneously.(168,169)
Continuous subcutaneous infusion using a portable battery-operated
syringe driver or other similar device was originally devised to
administer infusions of desferrioxamine to patients with thalassemia,
but was subsequently used to deliver diamorphine to patients with
advanced cancer who were unable to take oral drugs.(170)
This technique is now well established in palliative care and is used
to administer analgesics, antiemetics, anxiolytic sedatives, and
dexamethasone.
Ambulatory infusion devices vary in complexity, cost,
and ability to provide patient-controlled ‘rescue doses’ as an adjunct
to a continuous basal infusion. A variety of devices have been
employed, all designed to be lightweight and portable, and in one case
disposable. Opioids suitable for continuous subcutaneous infusion must
be soluble, well absorbed and non-irritant. Extensive experience has
been reported with morphine, diamorphine, hydromorphone, fentanyl, and
oxymorphone.(120,168,171) Methadone(110) and fentanyl appear to be relative irritants and are best avoided by this route.
Studies suggest that dosing with subcutaneous
administration can proceed in a manner identical to continuous
intravenous infusion: a post-operative study comparing patients who
received an identical dose of morphine by either intravenous or
subcutaneous infusion found no difference in blood levels,(172)
and a controlled study of hydromorphone calculated a bioavailability of
78 per cent for the subcutaneous route and observed that analgesic
outcome was identical during intravenous or subcutaneous infusion. To
maintain the comfort of an infusion site, the subcutaneous infusion
rate should not exceed 5 ml/h. Subcutaneous infusion has become the
first choice when parenteral analgesia is required in palliative care
patients.
Continuous intravenous infusion may be the most
appropriate way of delivering an opioid for patients with a
pre-existing implanted central line, when there is a need for infusion
of a large volume of solution, or when using methadone. If continuous
intravenous infusion must be continued on a long-term basis, a
permanent central venous port is recommended.
Continuous infusions of drug combinations may be
indicated when pain is accompanied by nausea, anxiety, or agitation. In
such cases an antiemetic, neuroleptic, or anxiolytic may be combined
with an opioid provided that it is non-irritant, miscible, and stable
in combined solution. As noted later in the text, a variety of
different combinations of drugs are commonly given by continuous
infusion.(173) However, the
stability/compatibility of many of these combinations is not known. The
compatibility of drug combinations is dependent on a number of factors,
including the types of drugs, the concentrations of drugs, the diluent
and temperature, and UV light. A database of compatible drug
combinations is now available on the internet (http://www.pallmed.net/).
Generally infusions should contain as few drugs as possible, preferably
no more than three. The absence of precipitation within a drug mixture
is not synonymous with compatibility between the drugs in that mixture.(174)
Epidural, intrathecal, and intraventricular administration (see also Chapter 8.2.6)
The discovery of opioid receptors in the dorsal horn of
the spinal cord led to the development of intraspinal opioid delivery
techniques. In general, they provide a longer duration of analgesia at
doses lower than required by systemic administration. The delivery of
low opioid doses near the sites of action in the spinal cord may
decrease supraspinally mediated adverse effects. Opioid selection for
intraspinal delivery is influenced by several factors. Hydrophilic
drugs, such as morphine and hydromorphone, have a prolonged half-life
in cerebrospinal fluid and significant rostral redistribution.(175)
Lipophilic opioids, such as fentanyl and sufentanil, have less rostral
redistribution and therefore less prolonged adverse effects if these
become a problem.
The addition of local anaesthetic such as bupivacaine to
an epidural or intrathecal opioid has been demonstrated to improve
analgesia without increasing toxicity.(176,177)
Unlike in acute post-operative pain, where a large volume of low
concentration local anaesthetic is used, in chronic cancer pain, a
small volume of high concentration of local anaesthetic is preferred
and can be mixed with an appropriate dose of a small volume of opioid.
The initial conversion of opioid dose from systemic subcutaneous diamorphine or morphine is:
- epidural—1/10 of systemic dose;
- intrathecal—1/10 of epidural dose.
Thus, if a patient were on 100 mg of subcutaneous
morphine or diamorphine/ day, the equivalent epidural dose would be 10
mg, and the equivalent intrathecal dose would be 1 mg/day.
The initial solution used for epidural infusion is usually:
- 9 ml 0.5 per cent bupivacaine;
- 150 µg clonidine;
- morphine or diamorphine dose according to individual patient requirements (as calculated above).
This gives a total volume of 10 ml infused over 24 h.
The initial solution used for intrathecal infusion is normally around 1/10 of the above, that is:
- 1 ml 0.5 per cent bupivacaine;
- 15 µg clonidine;
- morphine or diamorphine according to individual patient requirements.
Should there be a major problem with pump malfunction, and the whole dose is delivered as a bolus, this should not result in a major, life-threatening overdose.
There are no trials comparing the intrathecal and
epidural routes in cancer pain. The epidural route is generally
preferred because the techniques to accomplish long-term administration
are simpler. A combined analysis of adverse effects observed in
numerous trials of epidural or intrathecal administration suggests that
the risks associated with these techniques are similar (see Chapter 8.2.6).
The potential morbidity associated with these procedures emphasizes the
need for a well-trained clinician and long-term monitoring.
Limited experience suggests that the administration of
an opioid into the cerebral ventricles can provide long-term analgesia
in selected patients. This technique has been used for patients with
upper-body or head pain or with severe diffuse pain. Schedules have
included both intermittent injection via an Ommaya reservoir and
continual infusion using an implanted pump.
The indication for the spinal routes of administration
of opioid analgesics in palliative care patients is discussed in more
detail in Chapter 8.2.6.
Other routes and modes of administration
Transdermal
As previously described, fentanyl and buprenorphine are available in a transdermal formulation and their use is discussed above.
Sublingual
Sublingual absorption could potentially occur with any
opioid, but bioavailability is very poor with drugs that are not highly
lipophilic.(178,179) A
sublingual preparation of buprenorphine is available in some countries,
although not in the United States. Anecdotally, sublingual morphine has
also been reported to be effective; given the poor sublingual
absorption of
this drug, this efficacy may be related in part to swallowing of the dose. Both fentanyl and methadone are well absorbed sublingually, but no preparations are currently available for clinical use. Thus the sublingual approach has limited value owing to the lack of true sublingual formulations, poor absorption of most drugs, and the inability to deliver high doses or to prevent swallowing of the dose. Sublingual administration of an injectable formulation is occasionally used in patients requiring low doses of opioids who temporarily lose the option of oral dosing. Sublingual administration of fentanyl and related drugs (alfentanyl) has also been reported to be effective in the management of breakthrough pain.(180) The injectable formulation is used and is much cheaper than the oral transmucosal formulation of fentanyl (OTFC). However, it is not really a practical option for patients who are still mobile and at home for whom OTFC may work well and is more convenient to use.
P.330
this drug, this efficacy may be related in part to swallowing of the dose. Both fentanyl and methadone are well absorbed sublingually, but no preparations are currently available for clinical use. Thus the sublingual approach has limited value owing to the lack of true sublingual formulations, poor absorption of most drugs, and the inability to deliver high doses or to prevent swallowing of the dose. Sublingual administration of an injectable formulation is occasionally used in patients requiring low doses of opioids who temporarily lose the option of oral dosing. Sublingual administration of fentanyl and related drugs (alfentanyl) has also been reported to be effective in the management of breakthrough pain.(180) The injectable formulation is used and is much cheaper than the oral transmucosal formulation of fentanyl (OTFC). However, it is not really a practical option for patients who are still mobile and at home for whom OTFC may work well and is more convenient to use.
Oral transmucosal and nasal
The management of ‘breakthrough’ pain has been a topic of considerable recent interest(181)
partly stimulated by the development of OTFC and other novel approaches
to the administration of potent opioids. The nasal route may be
effective for a number of opioids(182) and
nasal diamorphine spray has been shown to provide effective pain relief
for children and teenagers presenting to emergency departments in acute
pain with clinical fractures.(183) This approach is also currently being investigated in adult cancer patients with troublesome acute episodic pain.
Topical
There are several case series and one very small
randomized controlled trial (in press) that examine the role of topical
morphine for local analgesia. The small amount of existing evidence
seems to point to a role in some situations, for example, cutaneous
ulcers or tumour with cutaneous inflammation. Doses of 10–40 mg of
morphine are used in simple gel, saline soaks, or local anaesthetic
gel.(184,185 and 186)
Changing the route of administration
As described above, when changing from the oral to
parenteral routes, or vice versa, an adjustment in dose is required to
avoid either toxic effects or a reduction in analgesia. The ratios of
oral to parenteral relative potency given in Table 3
are estimates and should not be taken as precise figures but used as
guidelines to achieve a roughly equianalgesic effect. There is
considerable variation between patients, and upward or downward
adjustment may then be required for individual patients. The slower
onset of analgesia after oral administration often requires some
adaptation on the part of a patient who is accustomed to the more rapid
onset seen after parenteral opioid. In some patients, the problems
associated with switching from the parenteral to the oral route of
opioid administration may need to be minimized by slowly reducing the
parenteral dose and increasing the oral dose over a 2–3-day period.
Usually, no dose adjustment is required when patients are switched from the subcutaneous to the intravenous route or vice versa.
Scheduling opioid administration
‘Around-the-clock’ dosing
To provide the patient with continuous relief by
preventing the pain from recurring, patients with continuous or
frequent pain are usually scheduled for ‘around-the-clock’ dosing.
However, clinical vigilance is required in patients with no previous
opioid exposure and those administered drugs with long half-lives. With
methadone, for example, delayed toxicity may develop as plasma
concentration rises slowly toward steady state levels.
Rescue doses
All patients who receive an around-the-clock opioid
regimen should also be offered a ‘rescue dose’, that is, a supplemental
dose given on an as-needed basis to treat pain that breaks through the
regular schedule.(30) The integration of
scheduled dosing with rescue doses provides a method for safe and
rational stepwise dose escalation and is applicable to all routes of
opioid administration. The rescue drug is typically identical to that
administered on a continuous basis, with the exception of transdermal
fentanyl and methadone; the use of an alternative short-half-life
opioid is recommended for the rescue dose when these drugs are used.
The frequency with which the rescue dose can be administered depends on
the time to peak effect for the drug and the route of administration.
Oral rescue doses can be offered up to every 60–90 min, and parenteral
rescue doses can be offered up to every 15–30 min. Clinical experience
suggests that the size of the rescue dose should be equivalent to
one-sixth of the 24-h baseline dose, that is, the same as the 4-hourly
dose of opioid. The magnitude of the rescue dose should be
individualized and some patients with low baseline pain but severe
exacerbations may require rescue doses that are substantially larger.
Scheduling with sustained-release formulations
Sustained-release formulations can reduce the
inconvenience associated with around-the-clock administration. These
formulations should not be used for rapid titration of the dose in
patients with severe pain. Sustained-release oral morphine sulfate and
oxycodone, and transdermal fentanyl are now widely used, and
sustained-release formulations of codeine, tramadol, and hydromorphone
have been introduced in various countries.
A normal-release formulation of a short-half-life opioid
(usually the same drug) is generally used as the rescue medication.
Sustained- and normal-release formulations of oral morphine are dose
equivalent; switching from one to the other is done on a
milligram-for-milligram basis after the daily dose requirement is
identified using a normal-release formulation.
As-needed dosing
In some limited situations, an as-needed dosing regimen
alone can be recommended. This type of dosing provides additional
safety during the initiation of opioid therapy in the opioid-naive
patient, particularly when rapid dose escalation is needed or a
long-half-life drug is administered. This technique is strongly
recommended when starting methadone therapy and for patients with acute
renal failure.
Patient-controlled analgesia
Patient-controlled analgesia is a technique of
parenteral drug administration in which the patient controls a pump
that delivers bolus doses of an analgesic according to parameters set
by the physician. Use of a patient-controlled analgesia device allows
the patient to titrate the opioid dose carefully to his or her
individual analgesic needs. Long-term patient-controlled analgesia in
cancer patients is accomplished via subcutaneous or intravenous routes
using an ambulatory infusion device.(187) The
more technologically advanced of these devices have programmable
variables, including infusion rate, rescue dose, and lock-out interval.
The option for bolus dosing is typically used in conjunction with
continuous opioid infusion. There is relatively little experience of
patient-controlled analgesia in chronic cancer pain; it is a technique
largely confined to the management of acute post-operative pain.
Dose selection and adjustment
Initial dose selection
A patient with severe pain that is not controlled with a
step 2 opioid- non-opioid combination in full dose should begin one of
the opioid agonists at a dose equivalent to 10 mg oral morphine
sulphate every 4 h.
Dose titration
Inadequate pain relief should be addressed by gradual
escalation of the opioid dose until adequate analgesia is reported or
intolerable side-effects (that cannot be managed by simple
interventions) supervene. Because analgesic response to opioids
increases linearly with the logarithm of the dose, dose escalations of
less than 30–50 per cent are not likely to improve analgesia
significantly. Clinical experience indicates that a dose increment of
this order of magnitude is safe and is large enough to observe a
meaningful change
in effects. In most cases, gradual dose escalation identifies a favourable balance between analgesia and side-effects which remains stable for a prolonged period. While doses can become extremely large during this process, the absolute dose is immaterial as long as the balance between analgesia and side-effects remains favourable. In a retrospective study of 100 patients with advanced cancer, the average daily opioid requirement was equivalent to 400–600 mg of intramuscular morphine, but approximately 10 per cent of patients required more than 2000 mg and one patient required over 30000 mg every 24 h. Other centres have generally reported lower doses. A median dose of 60 mg/day in one centre(188) and 120 mg/day in another.(189)
P.331
in effects. In most cases, gradual dose escalation identifies a favourable balance between analgesia and side-effects which remains stable for a prolonged period. While doses can become extremely large during this process, the absolute dose is immaterial as long as the balance between analgesia and side-effects remains favourable. In a retrospective study of 100 patients with advanced cancer, the average daily opioid requirement was equivalent to 400–600 mg of intramuscular morphine, but approximately 10 per cent of patients required more than 2000 mg and one patient required over 30000 mg every 24 h. Other centres have generally reported lower doses. A median dose of 60 mg/day in one centre(188) and 120 mg/day in another.(189)
A simple method of dose titration using oral morphine is
to prescribe a dose of immediate-release morphine every 4 h and the
same dose for rescue (for breakthrough pain).(30)
The rescue dose can be given as often as required (e.g. every hour) and
the total dose of morphine can be reviewed daily. The regular dose can
then be adjusted according to how many rescue doses have been given.
Rate of dose titration
The severity of the pain should determine the rate of
dose titration. Patients with very severe pain can be managed by
repeated parenteral dosing every 15–30 min until pain is partially
relieved when an oral dosing regimen should be started.
Tolerance
Patients vary greatly in the opioid dose required to
manage their pain. The need for escalating doses is a complex
phenomenon. Most patients reach a dose that remains constant for
prolonged periods. When the need for dose escalation arises, any of a
variety of distinct processes may be involved. Clinical experience
suggests that true pharmacological tolerance is a much less common
reason than disease progression or increasing psychological distress.
Changes in the pharmacokinetics of an analgesic drug could also be
implicated.
True pharmacological tolerance probably involves changes
at the receptor level, and in this situation continued drug
administration itself induces an attenuation of effect. Clinically,
tolerance to the non-analgesic effects of opioids appears to occur
commonly albeit at varying rates for different effects. For example,
tolerance to respiratory depression, somnolence, and nausea generally
develops rapidly, whereas tolerance to opioid-induced constipation
develops very slowly, if at all. Tolerance to these opioid side-effects
is not a clinical problem, and indeed is a desirable outcome that
allows effective dose titration to proceed.
From the clinical perspective, the concern is that
tolerance to the analgesic effect of the drug will develop and that
this will necessitate rapid dose escalation which may continue until
the drug is no longer useful. Induction of true analgesic tolerance
which could compromise the utility of treatment can only be said to
occur if a patient manifests a need for increasing opioid doses in the
absence of other factors (e.g. progressive disease) that would be
capable of explaining the increase in pain. Extensive clinical
experience suggests that most patients who require an escalation in
dose to manage increasing pain have demonstrable progression of disease.
This conclusion has two important implications: concern
about tolerance should not impede the use of opioids early in the
course of the disease, and worsening pain in a patient receiving a
stable dose of opioid should not be attributed to tolerance but taken
as presumptive evidence of disease progression or, less commonly,
increasing psychological distress.
Opioid pharmacokinetic factors that may influence drug dosing
Hepatic and renal impairment
The impact of hepatic and renal impairment on opioid
metabolism and excretion has been described previously. Neither hepatic
nor renal dysfunction is a contra-indication to the use of opioid
analgesics in cancer pain. Care is required, particularly in patients
with renal impairment, but most situations can be managed without
complex or exceptional measures. Opioids may exacerbate the central
nervous system signs and symptoms in patients with severe hepatic or
renal dysfunction. A high level of vigilance is required, and clinical
signs and symptoms are more important than biochemical data in
indicating appropriate intervention.
Drug interactions
The tricyclic antidepressants clomipramine and
amitriptyline may increase plasma morphine levels as measured by an
increase in bioavailability and the half-life of morphine in cancer
patients.(190) The concurrent administration of
drugs that induce the hepatic mixed function oxidase system can alter
the disposition of certain opioids. The metabolism of pethidine is
increased by phenobarbitone and phenytoin, and that of methadone is
increased by phenytoin and rifampicin. Methadone has also been reported
to induce its own metabolism.
The potential for additive side-effects and serious
toxicity from drug combinations must be recognized. The sedative effect
of an opioid may add to that produced by numerous other centrally
acting drugs such as anxiolytics, neuroleptics, and antidepressants.
Likewise, the constipating effects of opioids are probably worsened by
drugs with anticholinergic effects. A severe adverse reaction,
including excitation, hyperpyrexia, convulsions, and death, has been
reported after the administration of pethidine to patients treated with
a monoamine oxidase inhibitor.
Advanced age
It appears that all phases of pharmacokinetics are
affected by the ageing process. Absorption may be influenced by
decreases in gastric acid, intestinal blood flow, mucosal cell mass,
and intestinal motility. The clearance of morphine, fentanyl, and
nalbuphine is decreased in the elderly, and this age-related difference
in pharmacokinetics may partially explain the greater sensitivity of
older patients to therapeutic opioid doses compared with younger
patients. Pharmacodynamic responses may also be altered in older
patients. Increased receptor sensitivity and concurrent alterations in
mental status may account in part for the increased response shown by
elderly patients to opioid analgesics. In practice, reducing the dose
or lengthening the time interval between doses for the elderly patient
will minimize the development of serious adverse effects.
Children
The management of pain in children with opioid analgesics follows the same principles as described for the adult patient (see Chapter 9.1).
The oral and intravenous routes are commonly used to avoid repetitive
needle injections. Continuous subcutaneous infusion has also been used
in the terminally ill child. Individualization of doses and titration
to the needs of the child are essential.
Management of opioid adverse effects
Successful opioid therapy requires that the benefits of
analgesia clearly outweigh treatment-related adverse effects. This
requires understanding of adverse opioid effects and the strategies
used to prevent and manage them are essential skills for all involved
in cancer pain management.(191) The adverse effects that are frequently observed in patients receiving oral morphine and other opioids are summarized in Table 4.
The most common are sedation, constipation, and nausea and vomiting,
but there are other adverse effects including confusion,
hallucinations, nightmares, urinary retention, multifocal myoclonus,
dizziness, and dysphoria. The mechanisms that underlie these various
adverse effects, even the most common, are only partly understood and,
as discussed above, appear to depend upon a number of factors including
age, extent of disease and organ dysfunction, concurrent administration
of certain drugs, prior opioid exposure, and the route of drug
administration. Studies comparing the adverse effects of one opioid
analgesic with another in this population are lacking. Similarly,
controlled studies comparing the adverse effects produced by the same
opioid given by various routes of administration are also lacking.
Table 4 Common opioid-induced adverse effects | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
P.332
As a general rule, caution is required when using
opioids in patients in acute pain with impaired ventilation, bronchial
asthma, or raised intracranial pressure; the same caveats do not
usually limit dose titration in chronic cancer pain management.
Factors predictive of opioid adverse effects
Drug related
Overall, there is very little reproducible evidence
suggesting that any one opioid agonist has a substantially better
adverse effect profile than any other. Pethidine is not recommended in
the management of chronic cancer pain because of concerns regarding its
side-effect profile. Recent data from controlled studies indicate that
the transdermal administration of fentanyl is associated with a lesser
incidence of constipation than oral morphine.(145,192)
Route related
There is very limited evidence to suggest differences in
adverse effects associated with specific routes of systemic
administration. Compared to oral administration of morphine, small
studies have demonstrated less nausea and vomiting with rectal(193) and subcutaneous administration.(194)
As noted above, transdermal fentanyl appears to be associated with less
constipation than oral morphine. It is not clear whether this is a
route- or drug-related effect.
Patient related
For reasons that are not well explained, there is
striking interindividual variability in the sensitivity to adverse
effects from morphine and other opioid drugs. Genetic variability may
be at least part of the explanation in that it may influence both
therapeutic and unwanted effects.
Some of this variability is related to co-morbidity.
Ageing is associated with altered pharmakokinetics particularly
characterized by diminished clearance and volume of distribution. This
has been well described for morphine(195) and fentanyl.(196,197)
In a study of morphine in chronic cancer pain, overall elderly patients
required lower doses than their younger counterparts without exhibiting
an enhanced risk for opioid induced adverse effects.(198) In patients with impaired renal function there is delayed clearance of the metabolite M6G.(199) Anecdotally, high concentrations of M6G have been associated with toxicity,(76,200,201)
however, in a prospective study of patients with opioid-induced
delirium or myoclonus no relationship to renal function was observed.(202)
Other patient-related factors that may enhance the risk
of adverse effects include the co-administration of drugs which may
have cumulative toxicity or other concurrent co-morbidity (Table 5).
Table 5 Co-morbidity that may mimic opioid-induced adverse effects | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Opioid initiation and dose escalation
Some adverse effects appear transiently, after the
initiation of an opioid or after dose escalation and spontaneously
abate. This phenomenon has been well demonstrated in a prospective
study of morphine dose escalation and its effects on cognitive
function.(203) This study demonstrated that
cognitive impairment which was evident at the start of treatment with
morphine or when the dose was increased commonly improved after 7 days.
This phenomenon, though often described, has not been formally studied
with other adverse effects.
There is substantial variability in the dose response of
opioid adverse effects. A dose–response relationship is most commonly
evident with central
nervous system effects of sedation, cognitive impairment, hallucinations, myoclonus, and respiratory depression. Even among these, however, there is very substantial interindividual variability Additionally, as tolerance develops to some effects, the spectrum of adverse effects changes over time. Commonly, patients who have had prolonged opioid exposure have a lesser tendency to develop sedation or respiratory depression, and the predominant central nervous system effects become the neuroexcitatory ones of delirium and myoclonus. Gastrointestinal adverse effects generally have a weaker dose- response relationship. Some, like nausea and vomiting, are common with the initiation of therapy but are subsequently unpredictable with resolution among some patients and persistence among others. Constipation is virtually universal and demonstrates no consistent dose relationship.
P.333
nervous system effects of sedation, cognitive impairment, hallucinations, myoclonus, and respiratory depression. Even among these, however, there is very substantial interindividual variability Additionally, as tolerance develops to some effects, the spectrum of adverse effects changes over time. Commonly, patients who have had prolonged opioid exposure have a lesser tendency to develop sedation or respiratory depression, and the predominant central nervous system effects become the neuroexcitatory ones of delirium and myoclonus. Gastrointestinal adverse effects generally have a weaker dose- response relationship. Some, like nausea and vomiting, are common with the initiation of therapy but are subsequently unpredictable with resolution among some patients and persistence among others. Constipation is virtually universal and demonstrates no consistent dose relationship.
Differential diagnosis
Adverse symptoms in patients taking opioids are not
always caused by the opioid. Drug induced effects must be
differentiated from other causes and from drug interactions (Table 2).
Indeed, the appearance of a new adverse change in patient well-being
that occurs in the setting of stable opioid dosing is rarely caused by
the opioid, and an alternative explanation should be vigorously sought.
Strategies for management of opioid adverse effects
In general, four different approaches to the management of opioid adverse effects have been described:
- dose reduction of systemic opioid;
- specific therapy to reduce the adverse effect;
- opioid switch (or rotation);
- change in the route of administration.
Dose reduction of systemic opioid
Reducing the dose of administered opioid usually results
in a reduction in dose-related adverse effects. When patients have
well-controlled pain, gradual reduction in the opioid dose will often
result in the resolution of dose-related adverse effects whilst
preserving adequate pain relief.(204)
When opioid doses cannot be reduced without the loss of
pain control, reduction in dose must be accompanied by the addition of
an accompanying synergistic approach.
The addition of a non-opioid co-analgesic
The analgesia achieved from NSAIDs is additive and often synergistic with that achieved by opioids.(205,206,207 and 208)
The addition of an adjuvant analgesic that is appropriate to the pain syndrome and mechanism (see Chapter 8.2.5)
Adjuvant analgesics (see below) may be combined with
primary analgesics to improve the outcome for patients who cannot
otherwise attain an acceptable balance between pain relief and
side-effects.(209) There is great
interindividual variability in the response to all adjuvant analgesics
and, for many only limited benefit. Many of the adjuvant analgesics
have the potential to cause side-effects that may be additive to the
opioid-induced adverse effects that are already problematic. In
evaluating the utility of an adjuvant agent in a particular patient
setting, one must consider the likelihood of benefit, the risk of
adverse effects, the ease of administration, and patient convenience.
The application of a therapy targeting the cause of the pain
Specific antitumour therapies, such as radiotherapy,
chemotherapy, or surgery targeting the cause of cancer related pain can
provide substantial relief and thus reduce the need for opioid
analgesia. Radiotherapy is of proven benefit in the treatment of
painful bone metastases, epidural neoplasm and headache due to cerebral
metastases (see Chapter 8.1.2). In other
settings, there is a lack of well established supportive data, and the
use of radiotherapy is largely anecdotal. Despite a paucity of evidence
concerning the specific analgesic benefits of chemotherapy,(210,211)
there is a strong clinical impression that tumour shrinkage is
generally associated with relief of pain. Surgery may have a role in
the relief of symptoms caused by specific problems, such as obstruction
of a hollow viscus, unstable bony structures and compression of neural
tissues (Chapter 8.1.3 and Chapter 8.1.4).
The application of a regional anaesthetic or neuroablative intervention (see Chapter 8.2.6)
The results of the WHO analgesic ladder validation
studies suggest that 10–30 per cent of patients with cancer pain do not
achieve a satisfactory balance between relief and side-effects using
systemic pharmacotherapy alone without unacceptable drug toxicity.
Anaesthetic and neurosurgical techniques may reduce or eliminate the
requirement for systemically administered opioids to achieve adequate
analgesia. In general, regional analgesic techniques such as
intraspinal opioid and local anaesthetic administration or intrapleural
local anaesthetic administration are usually considered first because
they can achieve this end without compromising neurological integrity.
Neurodestructive procedures, however, are valuable in a small subset of
patients; and some of these procedures, such as coeliac plexus blockade
in patients with pancreatic cancer, may have a favourable enough risk
benefit ratio that early treatment is warranted.
Symptomatic therapy to reduce the adverse effects
Symptomatic drugs used to prevent or control opioid
adverse effects are commonly employed. With few exceptions, the
literature describing these approaches is anecdotal. Very few studies
have prospectively evaluated efficacy and no studies have evaluated the
toxicity of these approaches over the long term. In general, this
approach involves the addition of a new medication, adding to
medication burden and with the associated risks of additional or
different adverse effects or drug interactions.
Opioid switch (or rotation)
It has long been observed anecdotally that patients who
develop intolerable adverse effects with morphine while achieving
inadequate analgesia may sometimes benefit from switching to an
alternative oral opioid agonist.(212,213)
In recent years, the practice has focused particularly on the problems
of cognitive impairment and the possible relationship with toxic
metabolites of morphine and other opioids. The frequency with which
this is a problem and the suggested pathophysiology has been the
subject of some controversy(214,215) and some have advocated multiple switches if the first change in drug does not achieve or maintain the desired effect.(216,217,218 and 219)
Improvements in cognitive impairment, sedation, hallucinations, nausea,
vomiting, and myoclonus have been commonly reported. This approach
requires familiarity with a range of opioid agonists and with the use
of equianalgesic tables to convert doses when switching between
opioids. While this approach has the practical advantage of minimizing
polypharmacy, outcomes are variable and unpredictable. When switching
between opioids, even with prudent use of equianalgesic tables,
patients are at risk for under-or-over dosing by virtue of individual
sensitivities.
Dose adjustments when switching opioids
When patients are switched from one opioid analgesic to
another, lack of attention to the drug-dependent differences in opioid
dose may result in undermedication or overdose. In this setting
familiarity with the use of the equianalgesic dose table (Table 3)
is essential. For patients with good pain control, the starting dose of
the new drug should be reduced to 50–75 per cent of the equianalgesic
dose to account for incomplete cross-tolerance. However, if the patient
had inadequate pain control on the previous opioid, a smaller dose
reduction is used and the starting dose of the new drug can be usually
75–100 per cent of the equianalgesic dose. Clinical experience suggests
that additional caution is needed when the change is to methadone; a
reduction to 25–33 per cent of the equianalgesic dose is prudent. After
any change from one opioid to another, patients must be
monitored to assess the adequacy of analgesia and to detect the development of side-effects. Subsequent dose adjustments are usually necessary.
P.334
monitored to assess the adequacy of analgesia and to detect the development of side-effects. Subsequent dose adjustments are usually necessary.
This information has been gained empirically but is
based upon the concept that cross-tolerance is not complete among
opioids and conforms to our recognition that the relative potency of
some of the opioid analgesics may change with repetitive dosing,
particularly those opioids with a long plasma half-life.
The biological basis for the observed interindividual
variability in sensitivity to opioid analgesia and adverse effects is
multifactorial. Preclinical studies show that opioids can act on
different receptors or sub-type receptors(220)
and individual receptor profiles may influence the analgesia as well as
the side-effects. The genetic makeup of the individual plays an
important role in analgesia for some opioids(221) and similar phenomena may contribute to variability in adverse effect sensitivity.
Change in the route of systemic administration
There is no good evidence that efficacy is dependent on
route of administration: in general, morphine has equal efficacy if
given in appropriate dosage by oral, parenteral, or spinal routes.
However, there are limited data that indicate that some adverse
side-effects among patients receiving oral morphine can be relieved by
switching the route of admission to the subcutaneous route. In one
small study, this phenomenon was reported for nausea and vomiting,(194) in another there was less constipation, drowsiness, and nausea.(222)
Initial management of the patient receiving opioids who presents with adverse effects
Among patients receiving opioid analgesic therapy, there
are two key steps in the initial management of adverse effects. First,
the clinician must distinguish between morphine adverse effects and
co-morbidity or drug interactions, and deal with the latter
appropriately. In patients with advanced cancer, side-effects due to
drug combinations are common. The potential for additive side-effects
and serious toxicity from drug combinations must be recognized. The
sedative effect of an opioid may add to that produced by numerous other
centrally acting drugs, such as anxiolytics, neuroleptics, and
antidepressants.(223) Likewise, drugs with anticholinergic effects probably worsen the constipating effects of opioids.
If it seems that there is a true adverse effect of the
opioid, consideration should be given to reducing the opioid dose. If
the patient has good pain control, the morphine dose should be reduced
by 25 per cent.
Gastrointestinal side-effects
The gastrointestinal adverse effects of opioids are
common. In general, they are characterized by having a weak
dose–response relationship.
Constipation
Constipation is the most common adverse effect of chronic opioid therapy.(224) Laxative medications should be routinely prescribed prophylactically to most patients.
Nausea and vomiting
Opioids may produce nausea and vomiting through both
central and peripheral mechanisms. These drugs stimulate the medullary
chemoreceptor trigger zone, increase vestibular sensitivity, and have
effects on the gastrointestinal tract (including increased gastric
antral tone, diminished motility, and delayed gastric emptying). With
the initiation of opioid therapy, patients should be informed that
nausea can occur and that it is usually transitory and controllable.
Routine prophylactic administration of an antiemetic is not necessary,
except in patients with a history of severe opioid-induced nausea and
vomiting, but patients should have access to an antiemetic at the start
of therapy if the need for one arises. Anecdotally, the use of
prochlorperazine, metoclopromide, or haloperidol in low dose has
usually been sufficient.
Central nervous system side-effects
The central nervous system side-effects of opioids are
generally dose related. The specific pattern of central nervous system
adverse effects is influenced by individual patient factors, duration
of opioid exposure and dose.
Sedation
Initiation of opioid therapy or significant dose
escalation commonly induces sedation that persists until tolerance to
this effect develops, usually in days to weeks. It is useful to
forewarn patients of this potential, and thereby reduce anxiety and
encourage avoidance of activities, such as driving, that may be
dangerous if sedation occurs.(225) Some
patients have a persistent problem with sedation, particularly if other
confounding factors exist. These factors include the use of other
sedating drugs or co-existent diseases such as dementia, metabolic
encephalopathy, or brain metastases. Both dextroamphetamine and
methylphenidate have been used in the treatment of opioid-induced
sedation.(226) Treatment with methylphenidate
or dextroamphetamine is typically begun at 2.5–5 mg in the morning,
which is repeated at midday if necessary to maintain effects until
evening. Doses are then increased gradually if needed. Few patients
require more than 40 mg per day in divided doses. This approach is
relatively contra-indicated among patients with cardiac arrhythmias,
agitated delirium, paranoid personality, and past amphetamine abuse.
Confusion and delirium
Mild cognitive impairment is common following the
initiation of opioid therapy or dose increases. Similar to sedation,
however, pure opioid-induced encephalopathy appears to be transient in
most patients, persisting from days to a week or two. Although
persistent confusion attributable to the opioid alone occurs, the
aetiology of persistent delirium is usually related to the combined
effect of the opioid and other contributing factors, including
electrolyte disorders, neoplastic involvement of the central nervous
system, sepsis, vital organ failure, and hypoxaemia.(226) A stepwise approach to management (Table 6)
often culminates in a trial of a neuroleptic drug. Haloperidol in low
doses (0.5–1.0 mg PO or 0.25–0.5 mg intravenously or intramuscularly)
is most commonly recommended because of its efficacy and low incidence
of cardiovascular and anticholinergic effects.
Table 6 A stepwise approach to the management of confusion and delirium | |||
|---|---|---|---|
|
Respiratory depression
When sedation is used as a clinical indicator of central
nervous system toxicity and appropriate steps are taken, respiratory
depression is rare. When, however, it does occur it is always
accompanied by other signs of central nervous system depression,
including sedation and mental clouding. Respiratory compromise
accompanied by tachypnoea and anxiety is never a primary opioid event.
With repeated opioid administration, tolerance appears
to develop rapidly to the respiratory depressant effects of opioid
drugs and consequently clinically important respiratory depression is a
very rare event in the cancer patient whose opioid dose has been
titrated against pain.
The ability to tolerate high doses of opioids is also
related to the stimulus related effect of pain on respiration in a
manner that is balanced against the
depressant opioid effect. Opioid-induced respiratory depression can occur, however, if pain is suddenly eliminated (such as may occur following neurolytic procedures) and the opioid dose is not reduced.(227)
P.335
depressant opioid effect. Opioid-induced respiratory depression can occur, however, if pain is suddenly eliminated (such as may occur following neurolytic procedures) and the opioid dose is not reduced.(227)
When respiratory depression occurs in patients on
chronic opioid therapy, administration of the specific opioid
antagonist, naloxone, usually improves ventilation. This is true even
if the primary cause of the respiratory event was not the opioid
itself, but rather, an intercurrent cardiac or pulmonary process. A
response to naloxone, therefore, should not be taken as proof that the
event was due to the opioid alone and an evaluation for these other
processes should ensue.
Naloxone can precipitate a severe abstinence syndrome
and should be administered only if strongly indicated. If the patient
is bradypnoeic but readily arousable, and the peak plasma level of the
last opioid dose has already been reached, the opioid should be
withheld and the patient monitored until improved. If severe
hypoventilation occurs (regardless of the associated factors that may
be contributing to respiratory compromise), or the patient is
bradypnoeic and unrousable, naloxone should be administered. To reduce
the risk of severe withdrawal following a period of opioid
administration, dilute naloxone (1 : 10) should be used in doses
titrated to respiratory rate and level of consciousness. In the
comatose patient, it may be prudent to place an endotracheal tube to
prevent aspiration following administration of naloxone.
Multifocal myoclonus
All opioid analgesics can produce myoclonus. Mild and
infrequent myoclonus is common. In occasional patients, however,
myoclonus can be distressing or contribute to breakthrough pain that
occurs with the involuntary movement. If the dose cannot be reduced due
to persistent pain, consideration should be given to either switching
to an alternative opioid(216) or to symptomatic treatment with a benzodiazepine (particularly clonazepam or midazolam), dantrolene or an anticonvulsant.(226)
Other effects
Urinary retention
Opioid analgesics increase smooth muscle tone and can
occasionally cause bladder spasm or urinary retention (due to an
increase in sphincter tone). This is an infrequent problem that is
usually observed in elderly male patients. Tolerance can develop
rapidly but catheterization may be necessary to manage transient
problems.
Opioids and driving
The ability to continue driving is very important to
maintaining the quality of life of many patients with advanced cancer.
Many assume that they must stop driving whilst taking regular potent
opioid analgesics, but this is not necessarily so. The usual advice to
patients is that they should not drive or engage in other skilled
activities such as operating machinery when they first start on
morphine or a similar opioid, or when they increase the dose. However,
once the initial sedative effects have resolved and both the patient
and physician are confident that cognitive and psychomotor performance
are no longer impaired, driving and other similar activities may
restart.
This advice is based to a large extent on empirical
experience and there have been few objective data to substantiate it.
However, recent studies confirm, perhaps surprisingly, that morphine
produces little measurable impairment of cognitive and psychomotor
function,(228) particularly in patients receiving continuous treatment with stable doses.(229)
In one study, which used a battery of performance tests designed
specifically to assess functions related to driving ability, chronic
morphine use was associated with slower reaction times, more mistakes,
and a slowing in ability to process visual information and perform
motor sequences, but these changes were not statistically significant
compared with a control group of cancer patients not taking morphine.(225)
These data support the clinical impression that stable doses of
morphine are unlikely to cause substantial impairment of the
psychomotor skills required for driving, and allow us to continue to
advise patients to this effect.
‘Allergy’ and intolerance to morphine
Morphine and other opioids cause histamine release, and this is said to contribute to asthma or urticaria in allergic patients.(133,230,231) There is no published information on the incidence of this phenomenon and, in our experience, it is very rare.
However, it is not uncommon for patients to claim that
they are ‘allergic’ to morphine. This usually means that they have had
a bad experience with the drug but, on investigation, what they
describe are its common side-effects. There is no doubt that most
patients experience some adverse effects when they first start regular
morphine treatment. Most commonly this is sedation, nausea, and, less
often, vomiting. All patients must be warned about this and appropriate
measures must be taken, as described above. If patients are not warned
and experience unpleasant adverse effects they will be discouraged from
continuing with the drug, and if they do not understand what is going
on they may assume they that are ‘allergic’ to it.
The opioid-dependent patient: definitions and misconceptions
Addiction and substance abuse are social and medical
problems of pandemic proportions which are associated with major social
and human costs. Commonly, opioid drugs are the preferred substances of
abuse. This association, combined with the principle of non-maleficence
is the basis for concern regarding risk of addiction caused by the
medical use of opioids. In recent years, the relationship between the
medical use of opioids and the risk of addiction has been the focus of
policy makers, medical sociologists, and pain clinicians. Overall, this
body of research has demonstrated: (i) the risk of developing addictive
behaviours or substance abuse as a consequence of the medical use of
opioids is low; (ii) patient, family members, members of the health
care professions, and regulators commonly overestimate the risk of
addiction; (iii) patient, family members, members of the health care
professions, and regulators often confuse physical dependence and
addiction and; (iv) together, these concerns contribute substantially
to physician reluctance to prescribe opioids and patient reluctance to
use them.(232,233 and 234)
To understand these phenomena as they relate to opioid
treatment of cancer pain, it is useful first to present a concept that
might be called ‘therapeutic dependence’. Patients who require a
specific drug therapy to control a symptom or disease process are
clearly dependent on the therapeutic efficacy of the drugs in question.
Examples of this ‘therapeutic dependence’ include the requirements of
patients with congestive cardiac failure for cardiotonic and diuretic
medication or the reliance of insulin-dependent diabetics on insulin
therapy. In these patients, undermedication or withdrawal of treatment
would result in serious untoward consequences. Patients with chronic
cancer pain have an analogous relationship to their analgesic therapy.
This relationship may or may not be associated with the development of
physical dependence, but is virtually never associated with addiction.
Psychological dependence and ‘addiction’
The properties of the opioid analgesics that are most
likely to lead to their being misused are effects mediated in the
central nervous system. The term addiction refers to a psychological
and behavioural syndrome characterized by a continued craving for an
opioid drug to achieve a psychic effect (psychological dependence) and
associated aberrant drug-related behaviours, such as compulsive
drug-seeking, unsanctioned use or dose escalation, and use despite harm
to self or others. Addiction should be suspected if patients
demonstrate compulsive use, loss of control over drug use, and
continuing use despite harm. The term addiction should not be used to
describe physical dependence.
There is a common perception that opioid use, for any
reason, is associated with a high risk of iatrogenic psychological
dependence and that it is best avoided or minimized.(235)
This bias is largely derived from experience with patients suffering
from substance abuse disorder who use opioids in settings other than
pain. Many health care professionals and laypersons fail to distinguish
between patients with substance abuse disorder and psychologically well
patients with pain, and consequently overestimate the risk
of iatrogenic addiction. This skews evaluation of the therapeutic index of opioids, and impacts adversely on the likelihood of a clinician to prescribe opioids and on the patients’ compliance with an opioid prescription.
P.336
of iatrogenic addiction. This skews evaluation of the therapeutic index of opioids, and impacts adversely on the likelihood of a clinician to prescribe opioids and on the patients’ compliance with an opioid prescription.
Despite very extensive use of opioids in the management
of acute pain and cancer pain, and a growing experience in the use of
opioids in recurrent acute pain and chronic non-cancer pain, there have
been relatively few studies that have specifically addressed the risk
of iatrogenic addiction.
In the only reported prospective study among cancer
patients treated with morphine, Schug et al. identified one case of
substance abuse among 550 cancer patients who were treated for a total
of 22525 treatment days.(236) In a national
survey of burn centres, no cases of addiction were identified among
more than 10000 patients who were administered opioids for pain.(237)
The largest prospective study involved a mixed population of 11 882
patients treated with acute or chronic cancer pain in the hospital
setting. In this study only four cases of addiction could be identified
among patients with no history of addiction who received at least one
dose of an opioid for strong pain.(238)
Finally, among 2369 headache patients, most of whom had access to
opioid therapy, only three cases of addiction were identified.(239)
Physical dependence
Physical dependence is the term used to describe the
phenomenon of withdrawal when an opioid is abruptly discontinued or an
opioid antagonist is administered.(240) The
severity of withdrawal is a function of the dose and duration of
administration of the opioid just discontinued (i.e. the patient's
prior opioid exposure). The administration of an opioid antagonist to a
physically dependent individual produces an immediate precipitation of
the withdrawal syndrome. Patients who have received repeated doses of a
morphine-like agonist to the point where they are physically dependent
may experience an opioid withdrawal reaction when given a mixed
agonist–antagonist. It can be shown that prior exposure to a
morphine-like drug greatly increases a patient's sensitivity to the
antagonist component of a mixed agonist–antagonist. Therefore, when
used for chronic pain, the mixed agonist–antagonists should be tried
before prolonged administration of a morphine-like agonist is initiated.
The abrupt discontinuation of an opioid analgesic in a
patient with significant prior opioid experience will result in signs
and symptoms characteristic of the opioid withdrawal or abstinence
syndrome.(241) The onset of withdrawal is
characterized by the patient's report of feelings of anxiety,
nervousness and irritability, and alternating chills and hot flushes. A
prominent withdrawal sign is ‘wetness’ including salivation,
lacrimation, rhinorrhoea, sneezing, and sweating, as well as
gooseflesh. At the peak intensity of withdrawal patients may experience
nausea and vomiting, abdominal cramps, insomnia, and, rarely,
multifocal myoclonus. The time course of the withdrawal syndrome is a
function of the elimination half-life of the opioid on which the
patient has become dependent. Abstinence symptoms generally appear
within 6–12 h and reach a peak at 24–72 h following cessation of a
short-half-life drug such as morphine, while onset may be delayed for
36–48 h with methadone, which has a long half-life. Therefore, it is
important to emphasize that, even in a patient in whom pain has been
completely relieved by a procedure (e.g. a cordotomy), it is necessary
to decrease the opioid dose slowly to prevent withdrawal.
Experience indicates that the usual daily dose required
to prevent withdrawal is equal to 75 per cent of the previous daily
dose. Following this rule of thumb, doses can be gradually titrated
down until the drug is discontinued.
‘Pseudoaddiction’
Some cancer patients who continue to experience
unrelieved pain manifest intense concern about opioid availability and
drug-seeking behaviour that is reminiscent of addiction but ceases once
pain is relieved, often through opioid dose escalation. This behaviour
has been termed ‘pseudoaddiction’.(242) Pain
relief usually produced by dose escalation eliminates this aberrant
behaviour and distinguishes the patient from the true addict.
Misunderstanding of this phenomenon may lead the clinician
inappropriately to stigmatize the patient with the label ‘addict’,
which may compromise care and erode the doctor–patient relationship. In
the setting of unrelieved pain, the request for increases in drug doses
requires careful assessment, renewed efforts to manage pain, and
avoidance of stigmatizing labels.
Management of cancer pain in patients with a history of drug abuse
Patients with a history of abuse of opioid analgesics may develop cancer and severe pain.(243,244)
The management of such patients is, in principle, exactly the same as
that outlined in this chapter. Our approach is to maintain such
patients on oral medication if possible, even though they may require
very much larger doses than normal. If parenteral medication is
required, continuous subcutaneous infusion remains the mode of
administration of choice.
For patients receiving therapy for drug abuse, with or
without opioid maintenance therapy (e.g. methadone maintenance), it is
essential that the issues relating to the use of opioid analgesics for
pain management are discussed not only with the patient but also with
his or her family and drug abuse counsellors, so that the patient's
support group reaches a consensus on the utility and appropriateness of
analgesic therapy. An open and supportive approach, and the use of
concomitant psychotropic medication as appropriate, will aid the
effective management of these patients.(244)
In managing such patients, clinicians should be aware of
several common issues that may confound therapy. Mental clouding,
either as an effect of disease progression or as an iatrogenic adverse
effect, commonly raises concerns about the relapse or recurrence of
psychological dependence. A request for escalation of their opioid dose
may be generated by increased psychological stress rather than pain
alone. Aberrant drug-seeking behaviour such as acquisition of opioids
from multiple sources, ‘loss’ of prescribed drugs or prescriptions,
unsanctioned dose escalation, and prescription fraud must be recognized
as suggestive of true addiction and addressed openly as such. In all
cases, one clinician should be identified as responsible for pain
management, and these patients should be reviewed frequently.(244,245)
Conclusions
There have been many developments in our understanding
of opioid pharmacology, particularly at a molecular level, in the 10
years since the first edition of this textbook. Genomic research
promises further major advances in allowing us to understand many of
the clinical dilemmas surrounding the use of these drugs in patients
with pain. New formulations and increased availability of a wide range
of drugs have resulted in much greater sophistication in their use. But
the overarching advice remains the same. That optimal therapy for the
cancer patient with pain depends on a comprehensive assessment of his
or her pain, medical condition, and psychosocial status as well as an
understanding of the clinical pharmacology of analgesic drugs. Cancer
patients with pain vary greatly in their response to analgesics.
Genetic, pharmacokinetic and pharmacodynamic factors, as well as
psychological factors, will influence the effectiveness of an analgesic
in an individual patient. Through a process of repeated evaluation and
continuous review, analgesic therapy with opioids, non-opioids, and
adjuvant analgesics is individualized so that a favourable balance
between pain relief and adverse pharmacological effects is maintained.
References
1. World Health Organization.Cancer Pain Relief 2nd edn. Geneva: WHO, 1996.
2. Portenoy, R.K. and Lesage, P. (1999). Management of cancer pain. Lancet 353, 1695–700.
3. Lötsch, J.
et al. (2002). The polymorphism A118G of the human mu-opioid receptor
gene decreases the pupil constrictory effect of morphine-6-glucuronide
but not that of morphine. Pharmacogenetics 12, 3–9.
P.337
4. Hughes, J. and Kosterlitz, H.W. (1983). Introduction (to opioid peptides). British Medical Bulletin 39, 1–3.
5. Pasternak, G. (2001). The pharmacology of mu analgesics: from patients to genes. Neuroscientist 7, 220–31.
6. Reisine, T. and Pasternak, G. (1996). Opioid analgesics and antagonists. In Goodman and Gilman's Pharmacological Basis of Therapeutics 9th edn (ed. J.G. Hardman, L.E. Limbird, P.B. Molinoff, R.W. Ruddon, and A.G. Gilman), pp. 521–55. New York: McGraw-Hill.
7. Martin, W.R., Eades, C.G., Thompson, J.A., Huppler, R.E., and Gilbert, P.E. (1976). The effects of morphine and nalorphine-like drugs in the non-dependent and morphine-dependent spinal dog. Journal of Pharmacology and Experimental Therapeutics 197, 517–32.
8. Pasternak, G.W. (1993). Pharmacological mechanisms of opioid analgesics. Clinical Neuropharmacology 16, 1–18.
9. Sindrup, S.H.
et al. (1991). Codeine increases pain thresholds to copper vaper laser
stimuli in extensive but not poor metabolisers of sparteine. Clinical Pharmacology and Therapeutics 49, 686–93.
10. Inturrisi, C.E. et al. (1983). Evidence from opiate binding studies that heroin acts through its metabolites. Life Sciences 33, 773–6.
11. Heiskanen, T., Olkkola, K.T., and Kalso, K. (1998). Effects of blocking CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone. Clinical Pharmacology and Therapeutics 64, 603–11.
12. Christensen, C.B. (1993). The opioid receptor binding profiles of ketobemidone and morphine. Pharmacology and Toxicology 73, 344–5.
13. Raynor, K. et al. (1995). Characterization of the cloned human mu opioid receptor. Journal of Pharmacology and Experimental Therapeutics 272, 423–8.
14. Traynor, J.R. (1996). The mu-opioid receptor. Pain Reviews 3, 221–48.
15. Johnson, R.E. (1997). Review of US clinical trials of buprenorphine. Research and Clinical Forums 19, 17–23.
16. World Health Organization.Cancer Pain Relief. Geneva: WHO, 1986.
17. Ventafridda, V., Tamburini, M., Caraceni, A., DeConno, F., and Naldi, F. (1987). A validation study of the WHO method for cancer pain relief. Cancer 59, 851–6.
18. Takeda, F. (1990). Japan's WHO cancer pain relief program. In Advances in Pain Research and Therapy Vol. 16 (ed. K.M. Foley, J.J. Bonica, V. Ventafridda, and M.V. Callaway), pp. 475–83. New York: Raven Press.
19. Walker, V.A. et al. (1988). Evaluation of WHO analgesic guidelines for cancer pain in a hospital-based palliative care unit. Journal of Pain and Symptom Management 3, 145–9.
20. Goisis, A. et al. (1989). Application of a WHO protocol on medical therapy for oncologic pain in an internal medicine hospital. Tumori 75, 470–2.
21. Schug, S.A. et al. (1990). Cancer pain management according to WHO analgesic guidelines. Journal of Pain and Symptom Management 5, 27–32.
22. Zech, D.F.J., Grond, S., Lynch, J., Hertel, D., and Lehman, K.A. (1995). Validation of World Health Organization guidelines for cancer pain relief. A 10-year prospective study. Pain 63, 65–76.
23. Mercadante, S. (1999). Pain treatment and outcomes for patients with advanced cancer who receive follow-up care at home. Cancer 85, 1849–58.
24. McQuay, H.J. and Moore, R.A. (1997). Antidepressants and chronic pain. British Medical Journal 314, 763–4.
25. Kalso, E. et al. (1998). Systemic local-anaesthetic-type drugs in chronic pain: a systematic review. European Journal of Pain 2, 3–14.
26. Jadad, A.R. and Browman, G.P. (1995). The WHO analgesic ladder for cancer pain management. Journal of the American Medical Association 274, 1870–3.
27. Benedetti, C. et al. (2000). NCCN Practice Guidelines for cancer pain. Oncology 14, 135–50.
28. Cleary, J.F. (2000). Cancer pain management. Cancer Control 7, 120–31.
29. Walsh, D. (2000). Pharmacological management of cancer pain. Seminars in Oncology 27, 45–63.
30. Hanks, G.W. et al. (2001). Morphine and alternative opioids in cancer pain: the EAPC recommendations. British Journal of Cancer 84, 587–93.
31. Agency for Health Care Policy and Research: Cancer Pain Management Panel. Management of Cancer Pain. Clinical Practice Guideline 9. Washington DC: US Department of Health and Human Services, 1994.
32. Sindrup, S.H. and Brosen, K. (1995). The pharmacogenetics of codeine hypoalgesia. Pharmacogenetics 5, 335–46.
33. Poulsen, L.
et al. (1996). Codeine and morphine in extensive and poor metabolizers
of sparteine: pharmacokinetics, analgesic effect and side effects. European Journal of Clinical Pharmacology 51, 289–95.
34. Persson, K., Hammarlund-Udenaes, M., Mortimer, O., and Rane, A. (1992). The postoperative pharmacokinetics of codeine. European Journal of Clinical Pharmacology 42, 663–6.
35. Vree, T.B. and Verwey-van Wissen, C.P. (1992). Pharmacokinetics and metabolism of codeine in humans. Biopharmaceutics and Drug Disposition 13, 445–60.
36. de Crean, A.J.M., Di Guiulio, G., Lampe-Schoenmaeckers, A.J.E.M., Kessels, A.G.H., and Kleijnen, J. (1996). Analgesic efficacy and safety of paracetamol–codeine combinations versus paracetamol alone: a systematic review. British Medical Journal 313, 321–5.
37. Moore, A., Collins, S., Carroll, D., and McQuay, H. (1997). Paracetamol with and without codeine in acute pain: a quantitative systematic review. Pain 70, 193–201.
38. Rowell, F.J., Seymour, R.A., and Rawlins, M.D. (1983). Pharmacokinetics of intravenous and oral dihydrocodeine and its acid metabolites. European Journal of Clinical Pharmacology 25, 419–24.
39. Barnes, J.N. and Goodwin, F.J. (1983). Dihydrocodeine narcosis in renal failure. British Medical Journal 286, 438–9.
40. Barnes, J.N., Williams, A.J., Tomson, M.J.F., Toseland, P.A., and Goodwin, F.J. (1985). Dihydrocodeine in renal failure: further evidence for an important role of the kidney in the handling of opioid drugs. British Medical Journal 290, 740–2.
41. Crome, P., Gain, R., Ghurye, R., and Flanagan, R.J.
(1984). Pharmacokinetics of dextropropopoxyphene and
nordextropropoxyphene in elderly hospital patients after single and
multiple doses of Distalgesic. Preliminary analysis of results. Human Toxicology 3, 41S–8S.
42. Inturrisi, C.E., Colburn, W.A., Vereby, K., Dayton, H.E., Woody, G.E., and O'Brien, C.P. (1982). Propoxyphene and norpropoxyphene kinetics after single and repeated doses of propoxyphene. Clinical Pharmacology and Therapeutics 31, 157–67.
43. Beaver, W.T. (1984). Analgesic efficacy of dextropropoxyphene and dextropropoxyphene-containing combinations: a review. Human Toxicology 3, 191S–220S.
44. Li Wan Po, A. and Zhang, W.Y. (1997). Systematic overview of co-proxamol to assess analgesic effects of addition of dextropropoxyphene to paracetamol. British Medical Journal 315, 1565–71.
45. Perrier, D. and Gibaldi, M. (1972). Influence of first-pass effect on the systemic availability of propoxyphene. Journal of Clinical Pharmacology 12, 449–52.
46. Rosenberg, W.M. et al. (1993). Dextropropoxyphene induced hepatotoxicity: a report of nine cases. Journal of Hepatology 19, 470–4.
47. Hantson, P. et al. (1995). Adverse cardiac manifestations following dextropropoxyphene overdose: can naloxone be helpful? Annals of Emergency Medicine 25, 263–6.
48. Pippenger, C.E. (1987). Clinically significant carbamazepine drug interactions: an overview. Epilepsia 28 (Suppl. 3), S71–6.
49. Justice, J.L. and Kline, S.S. (1988). Analgesics and warfarin. A case that brings up questions and cautions. Postgraduate Medicine 83, 217–18, 220-.
50. Whittington, R.M. (1984). Dextropropoxyphene deaths: coroner's report. Human Toxicology 3 (Suppl.), 175S–85S.
51. Leow, K.P.
et al. (1995). Pharmacokinetics and pharmacodynamics of oxycodone when
given intravenously and rectally to adult patients with cancer pain. Anesthesia and Analgesia 80, 296–302.
52. Kalso, E., Poyhia, R., Onnela, P., Linko, K., Tigerstedt, I., and Tammisto, T. (1991). Intravenous morphine and oxycodone for pain after abdominal surgery. Acta Anaesthesiologica Scandinavica 35, 642–6.
53. Glare, P.A. and Walsh, T.D. (1993). Dose-ranging study of oxycodone for chronic pain in advanced cancer. Journal of Clinical Oncology 11, 973–8.
54. Heiskanen, T. and Kalso, E. (1997). Controlled-release oxycodone and morphine in cancer related pain. Pain 73, 37–45.
P.338
55. Hanks, G.W. and Hawkins, C. (2000). Agreeing a gold standard in the management of cancer pain: the role of opioids. In The Effective Management of Cancer Pain (ed. R. Hillier, I. Finlay, J. Welsh, and A. Miles), pp. 57–77. London: Aesculapius Medical Press.
56. Raffa, R.B., Friderichs, E., Reimann, W., Shank, R.P., Codd, E.E., and Vaught, J.L.
(1992). Opioid and non-opioid components independently contribute to
the mechanism of action of tramadol, an ‘atypical’ opioid analgesic. Journal of Pharmacology and Experimental Therapeutics 260, 275–85.
57. Lee, C.R.
et al. (1993). Tramadol. A preliminary review of its pharmacodynamic
and pharmacokinetic properties, and therapeutic potential in acute and
chronic pain states. Drugs 46, 313–40.
58. Wilder Smith, C.H., Schimke, J., Osterwalder, B., and Senn, H.J. (1994). Oral tramadol, a mu-opioid agonist and monoamine reuptake-blocker, and morphine for strong cancer-related pain. Annals of Oncology 5, 141–6.
59. Grond, S. et al. (1999). High-dose tramadol in comparison to low-dose morphine for cancer pain relief. Journal of Pain and Symptom Management 18, 174–9.
60. Petzke, F. et al. (2001). Slow-release tramadol for treatment of chronic malignant pain—an open multicenter trial. Supportive Care in Cancer 9, 48–54.
61. Radbruch, L. et al. (1996). A risk–benefit assessment of tramadol in the management of pain. Drug Safety 15, 8–29.
62. McEvoy, A.
et al. (1996). Comparison of diclofenac sodium and morphine sulphate
for postoperative analgesia after day case inguinal hernia surgery. Annals of the Royal College of Surgeons of England 78, 363–6.
63. St Charles, C.S. et al. (1997). A comparison of ibuprofen versus acetaminophen with codeine in the young tonsillectomy patient. Otolaryngology—Head and Neck Surgery 117, 76–82.
64. Innes, G.D. et al. (1998). Ketorolac versus acetaminophen-codeine in the emergency department treatment of acute low back pain. Journal of Emergency Medicine 16, 549–56.
65. De Conno, F.
et al. (1991). A clinical study on the use of codeine, oxycodone,
dextropropoxyphene, buprenorphine, and pentazocine in cancer pain. Journal of Pain and Symptom Management 6, 423–7.
66. Kopp, M. (1994). Buprenorphine transdermal system (TTS) delivery rate 35 mcg/h in an open long-term study with chronic patients. In 3rd Congress of the European Federation of IASP Chapter Nice, France. September 2000. Abstracts.
67. Benyhe, S. (1994). Morphine: new aspects in the study of an ancient compound. Life Sciences 55, 969–79.
68. Sawe, J., Dahlstrom, B., Paalzow, L., and Rane, A. (1981). Morphine kinetics in cancer patients. Clinical Pharmacology and Therapeutics 30, 629–35.
69. Hoskin, P.J., Hanks, G.W., Aherne, G.W., Chapman, D., Littleton, P., and Filshie, J.
(1989). The bioavailability and pharmacokinetics of morphine after
intravenous, oral and buccal administration in healthy volunteers. British Journal of Clinical Pharmacology 27, 499–505.
70. Gourlay, G.K., Plummer, J.L., Cherry, D.A., and Purser, T. (1991). The reproducibility of bioavailability of oral morphine from solution under fed and fasted conditions. Journal of Pain and Symptom Management 6, 431–6.
71. Sawe, J., Svensson, J.O., and Rane, A. (1983). Morphine metabolism in cancer patients on increasing oral doses—no evidence for autoinduction or dose dependence. British Journal of Clinical Pharmacology 16, 85–93.
72. Max, M.B., Inturrisi, C.E., Kaiko, R.F., Grabinski, P.Y., Li, C.H., and Foley, K.M. (1985). Epidural and inthrathecal opiates: cerebrospinal fluid and plasma profiles in patients with chronic cancer pain. Clinical Pharmacology and Therapeutics 38, 631–41.
73. Paul, D., Standifer, K.M., Inturrisi, C.E., and Pasternak, G.W. (1989). Pharmacological characterization of morphine-6-beta-glucuronide, a very potent morphine metabolite. Journal of Pharmacology and Experimental Therapeutics 251, 477–83.
74. Shimomura, K. et al. (1971). Analgesic effect of morphine glucuronides. Tohoku Journal of Experimental Medicine 105, 45–52.
75. Pasternak, G.W., Bodnar, R.J., Clark, J.A., and Inturrisi, C.E. (1987). Morphine-6-glucuronide, a potent mu agonist. Life Sciences 41, 2845–9.
76. Osborne, J.R., Joel, S.P., and Slevin, M.L. (1986). Morphine intoxication in renal failure: the role of morphine-6-glucuronide. British Medical Journal 292, 1548–9.
77. Osborne, R., Thompson, P., Joel, S., Trew, D., Patel, N., and Slevin, M.L. (1992). The analgesic activity of morphine-6-glucuronide. British Journal of Clinical Pharmacology 34, 130–8.
78. Portenoy, R.K., Thaler, H.T., Inturrisi, C.E., Friedlander-Klar, H., and Foley, K.M.
(1992). The metabolite, morphine-6-glucuronide, contributes to the
analgesia produced by morphine infusion in pain patients with normal
renal function. Clinical Pharmacology and Therapeutics 51, 422–31.
79. Portenoy, R.K.
et al. (1991). Plasma morphine and morphine-6-glucuronide during
chronic morphine therapy for cancer pain: plasma profiles, steady state
concentrations and the consequences of renal failure. Pain 47, 13–19.
80. D'Honneur, G., Gilton, A., Sandouk, P., Scherrmann, J.M., and Duvaldestin, P.
(1994). Plasma and cerebrospinal fluid concentrations of morphine and
morphine glucuronides after oral morphine. The influence of renal
failure. Anesthesiology 81, 87–93.
81. Lehmann, K.A. and Zech, D. (1993). Morphine-6-glucuronide a pharmacologically active morphine metabolite: a review of the literature. European Journal of Pain 12, 28–35.
82. Peat, S.J., Hanna, M.H., Woodham, M., Knibb, A.A., and Ponte, J. (1991). Morphine-6-glucuronide: effects on ventilation in normal volunteers. Pain 45, 101–4.
83. Penson, R.T., Joel, S.P., Bakhshi, K., Clark, S.J., Langford, R.M., and Slevin, M.L. (2000). Randomised placebo-controlled trial of the activity of the morphine glucuronides. Clinical Pharmacology and Therapeutics 68, 667–76.
84. Penson, R.T., Joel, S.P., Roberts, M., Gloyne, A., Beckwith, S., and Slevin, M.L. (2000). The bioavailability and pharmacokinetics of subcutaneous, nebulized and oral morphine-6-glucuronide. British Journal of Clinical Pharmacology 53, 347–54.
85. Hanks, G.W. (1991). Morphine pharmacokinetics and analgesia after oral administration. Postgraduate Medical Journal 67 (Suppl. 2), S60–3.
86. Smith, M.T., Watt, J.A., and Cramond, T. (1990). Morphine-3-glucuronide—a potent antagonist of morphine analgesia. Life Sciences 47, 579–85.
87. Gong, Q.-L., Hedner, J., Bjorkman, R., and Hedner, T.
(1992). Morphine-3-glucuronide may functionally antagonise
morphine-6-glucuronide induced antinociception and ventilatory
depression in the rat. Pain 48, 249–55.
88. Yaksh, T.L. and Harty, G.J. (1987). Pharmacology of the allodynia in rats evoked by high dose intrathecal morphine. Journal of Pharmacology and Experimental Therapeutics 244, 501–7.
89. Labella, F.S., Pinsky, C., and Havlicek, V. (1979). Morphine derivatives with diminished opiate receptor potency show enhanced central excitatory activity. Brain Research 174, 263–71.
90. Hewett, K., Dickenson, A.H., and McQuay, H.J.
(1993). Lack of effect of morphine-3-glucuronide on the spinal
antinociceptive action of morphine in the rat: an electrophysicological
study. Pain 53, 59–63.
91. Penson, R.T., Joel, S.P., Clark, S., Gloyne, A., and Slevin, M.L. (2001). Limited phase I study of morphine-3-glucuronide. Journal of Pharmaceutical Sciences 90, 1810–16.
92. Houde, R.W., Wallenstein, S., and Beaver, W.T. (1965). Clinical measurement of pain. In Analgesics (ed. G. Stevens), pp. 75–122. New York: Academic Press.
93. Hanks, G.W., Hoskin, P.J., Aherne, G.W., Turner, P., and Poulain, P. (1987). Explanation for potency of repeated oral doses of morphine? Lancet ii, 723–5.
94. Twycross, R.G. (1988). The therapeutic equivalence of oral and subcutaneous/ intramuscular morphine sulphate in cancer patients. Journal of Palliative Care 2, 67–8.
95. Kaiko, R.F. (1986). Commentary: equianalgesic dose ratio of intramuscular/oral morphine, 1 : 6 versus 1 : 3. In Advances in Pain Research and Therapy Vol. 8 (ed. K.M. Foley and C.E. Inturrisi), pp. 87–93. New York: Raven Press.
96. Hanks, G.W. (1989). Controlled-release morphine (MST Contin) in advanced cancer: the European experience. Cancer 623, 2378–82.
97. Forman, W.B., Portenoy, R.K., Yanagihara, R.H., Hunt, C., Kush, R., and Shepard, K. (1993). A novel morphine sulphate preparation: clinical trial of a controlled-release morphine suspension in cancer pain. Palliative Medicine 7, 301–6.
P.339
98. Savarese, J.J., Goldenheim, P.D., Thomas, G.B., and Kaiko, R.F. (1986). Steady-state pharmacokinetics of controlled release oral morphine sulphate in healthy subjects. Clinical Pharmacokinetics 11, 505–10.
99. Poulain, P. et al. (1988). Relative bioavailability of controlled release morphine tablets (MST Continus) in cancer patients. British Journal of Anaesthesia 61, 569–74.
100. Gourlay, G.K., Cherry, D.A., Onley, M.M., Tordoff, S.G., Conn, D.A., Hood, G.M., and Plummer, J.L.
(1997). Pharmacokinetics and pharmacodynamics of 24 hourly Kapanol
compared to 12 hourly MST Contin in the treatment of severe cancer
pain. Pain 69, 295–302.
101. Inturrisi, C.E., Max, M.B., Foley, K.M., Schultz, M., Shin, S.-U., and Houde, R.W. (1984). The pharmacokinetics of heroin in patients with chronic pain. New England Journal of Medicine 210, 1213–17.
102. Pasternak, G.W. and Standifer, K.M.
(1995). Mapping of opioid receptors using antisense
oligodeoxynucleotides: correlating their molecular biology and
pharmacology. Trends in Pharmacological Sciences 16, 334–50.
103. Kaiko, R.F., Wallenstein, S.L., Rogers, A.G., Grabinski, P.Y., and Houde, R.W. (1981). Analgesic and mood effects of heroin and morphine in cancer patients with postoperative pain. New England Journal of Medicine 304, 1501–5.
104. Ripamonti, C. et al. (1997). An update on the clinical use of methadone for cancer pain. Pain 70, 109–15.
105. Davis, M.P. and Walsh, D.
(2001). Methadone for relief of cancer pain: a review of
pharmacokinetics, pharmacodynamics, drug interactions and protocols of
administration. Supportive Care in Cancer 9, 73–83.
106. Grochow, L., Sheidler, V., Grossman, S., Green, L., and Enterline, J.
(1989). Does intravenous methadone provide longer lasting analgesia
than intravenous morphine? A randomized double-blind study. Pain 38, 151–7.
107. Sawe, J. et al. (1981). Patient-controlled dose regimen of methadone for chronic cancer pain. British Medical Journal 282, 771–3.
108. Mercadante, S. et al. (1996). Patient-controlled analgesia with oral methadone in cancer pain: preliminary report. Annals of Oncology 7, 613–17.
109. Mathew, P. and Storey, P. (1999). Subcutaneous methadone in terminally ill patients: manageable local toxicity. Journal of Pain and Symptom Management 18, 49–52.
110. Bruera, E., Fainsinger, R., Moore, M., Thibault, R., Spoldi, E., and Ventafridda, V. (1991). Local toxicity with subcutaneous methadone. Experience of two centers. Pain 45, 141–5.
111. Ripamonti, C.
et al. (1998). Equianalgesic dose/ratio between methadone and other
opioid agonists in cancer pain: comparison of two clinical experiences.
Annals of Oncology 9, 79–83.
112. Szeto, H.H., Inturrisi, C.E., Houde, R., Saal, R., Cheigh, J., and Reidenberg, M.M. (1977). Accumulation of normeperidine an active metabolite of meperidine, in patients with renal failure or cancer. Annals of Internal Medicine 86, 738–41.
113. Eisendrath, S.J., Goldman, B., Douglas, J., Dimatteo, L., and Van, D.C. (1987). Meperidine-induced delirium. American Journal of Psychiatry 144, 1062–5.
114. Umans, J.G. and Inturrisi, C.E. (1982). Antinociceptive activity and toxicity of meperidine and normeperidine in mice. Journal of Pharmacology and Experimental Therapeutics 223, 203–6.
115. Sporer, K.A. (1995). The serotonin syndrome. Implicated drugs, pathophysiology and management. Drug Safety 13, 94–104.
116. Kaiko, R.F. et al. (1983). Central nervous system excitatory effects of meperidine in cancer patients. Annals of Neurology 13, 180–5.
117. Agency for Health Care Policy and Research: Acute Pain Management Panel. Acute Pain Management: Operative or Medical Procedures and Trauma. Washington DC: US Department of Health and Human Services, 1992.
118. Houde, R.W. (1986). Clinical analgesic studies of hydromorphone. In Advances in Pain Research and Therapy Vol. 8 (ed. K.M. Foley and C.E. Inturrisi), pp. 129–36. New York: Raven Press.
119. Sarhill, N. et al. (2001). Hydromorphone: pharmacology and clinical applications in cancer patients. Supportive Care in Cancer 9, 84–96.
120. Moulin, D.E., Kreeft, J.H., Murray, P.N., and Bouquillon, A.I. (1991). Comparison of continuous subcutaneous and intravenous hydromorphone infusions for management of cancer pain. Lancet 337, 465–8.
121. Hays, H.
et al. (1994). Comparative clinical efficacy and safety of immediate
release and controlled release hydromorphone for chronic severe pain. Cancer 74, 1808–16.
122. Brose, W.G., Tanalian, D.L., Brodsky, J.B., Mark, J.B.D., and Cousins, M.J. (1991). CSF and blood pharmacokinetics of hydromorphone and morphine following lumbar epidural administration. Pain 45, 11–17.
123. Collins, J.J.
et al. (1996). Patient-controlled analgesia for mucositis pain in
children: a three-period crossover study comparing morphine and
hydromorphone. Journal of Pediatrics 129, 722–8.
124. Lawlor, P. et al. (1997). Dose ratio between morphine and hydromorphone in patients with cancer pain: a retrospective study. Pain 72, 79–85.
125. Dixon, R., Crews, T., Inturrisi, C.E., and Foley, K.M. (1983). Levorphanol: pharmacokinetics and steady-state plasma concentrations in patients with pain. Research Communications in Chemistry, Pathology and Pharmacology 41, 3–17.
126. Wallenstein, S.L., Rogers, A.G., Kaiko, R.F., and Houde, R.W. (1986). Clinical analgesic studies of levorphanol in acute and chronic cancer pain. In Advances in Pain Research and Therapy Vol 8 (ed. K.M. Foley and C.E. Inturrisi), pp. 211–15. New York: Raven Press.
127. Moulin, D.E., Ling, G.S., and Pasternak, G.W. (1988). Unidirectional cross tolerance between morphine and levorphanol in the rat. Pain 33, 233–9.
128. Kalso, E. and Vainio, A. (1990). Morphine and oxycodone hydrochloride in the management of cancer pain. Clinical Pharmacology and Therapeutics 47, 639–46.
129. Poyhia, R., Vainio, A., and Kalso, E. (1993). A review of oxycodone's clinical pharmacokinetics and pharmacodynamics. Journal of Pain and Symptom Management 8, 63–7.
130. Kaiko, R.F. et al. (1996). Pharmacokinetic–pharmacodynamic relationships of controlled-release oxycodone. Clinical Pharmacology and Therapeutics 59, 52–61.
131. Salzman, R.T.
et al. (1999). Can a controlled-release oral dose form of oxycodone be
used as readily as an immediate-release form for the purpose of
titrating to stable pain control? Journal of Pain and Symptom Management 18, 271–9.
132. Eddy, N.B. and Lee, L.E. (1959). The analgesic equivalence and relative side action liability of oxymorphone. Journal of Pharmacology and Experimental Therapeutics 125, 116–21.
133. Hermens, J.M., Hanifin, J.M., and Hirshman, C.A. (1985). Comparison of histamine release in human skin mast cells by morphine, fentanyl and oxymorphone. Anesthesiology 62, 124–9.
134. Rogers, A. (1991). Considering histamine release in prescribing opioid analgesics. Journal of Pain and Symptom Management 6, 44–5.
135. Yeadon, M. and Kitchen, I.
(1988). Comparative binding of mu and delta selective ligands in whole
brain and pons/medulla homogenates from rat: affinity profiles of
fentanyl derivatives. Neuropharmacology 27, 345–8.
136. Hess, R., Steibler, G., and Herz, A. (1972). Pharmacokinetics of fentanyl in man and the rabbit. European Journal of Clinical Pharmacology 4, 135–41.
137. Mather, L.E. (1983). Clinical pharmacokinetics of fentanyl and its newer derivatives. Clinical Pharmacokinetics 8, 422–46.
138. Lehmann, K.A. and Zech, D. (1992). Transdermal fentanyl: clinical pharmacology. Journal of Pain and Symptom Management 7 (Suppl.), S8–16.
139. Varvel, J.R., Shafer, S.L., Hwang, S.S., Coen, P.A., and Stanski, D.R. (1989). Absorption characteristics of transdermally administered fentanyl. Anesthesiology 70, 928–34.
140. Southam, M.A. (1995). Transdermal fentanyl therapy: system design, pharmacokinetics and efficacy. Anti-Cancer Drugs 6 (Suppl. 3), 26–34.
141. Portenoy, R.K. et al. (1993). Transdermal fentanyl for cancer pain: repeated dose pharmacokinetics. Anesthesiology 78, 36–43.
142. Jeal, W. and Benfield, P. (1997). Transdermal fentanyl. A review of its pharmacological properties and therapeutic efficacy in pain control. Drugs 53, 109–38.
143. Megens, A., Artois, K., and Vermeire, J. (1998). Comparison of the analgesic and intestinal effects of fentanyl and morphine in rats. Journal of Pain and Symptom Management 15, 253–7.
144. Haazen, L., Noorduin, H., and Megens, A. (1999). The constipation-inducing potential of morphine and transdermal fentanyl. European Journal of Pain 3 (Suppl.), 9–15.
145. Payne, R. et al. (1998). Quality of life and cancer pain: satisfaction and side effects with transdermal fentanyl versus oral morphine. Journal of Clinical Oncology 16, 1588–93.
P.340
146. Fine, P.G. et al. (1991). An open label study of oral transmucosal fentanyl citrate (OTFC) for the treatment of breakthrough cancer pain. Pain 45, 149–53.
147. Farrar, J.T.
et al. (1998). Oral transmucosal fentanyl citrate: randomized,
double-blinded, placebo-controlled trial for treatment of breakthrough
pain in cancer patients. Journal of the National Cancer Institute 90, 611–16.
148. Christie, J.M.
et al. (1998). Dose-titration, multicenter study of oral transmucosal
fentanyl citrate for the treatment of breakthrough pain in cancer
patients using transdermal fentanyl for persistent pain. Journal of Clinical Oncology 16, 3238–45.
149. Egan, T.D. et al. (2000). Multiple dose pharmacokinetics of oral transmucosal fentanyl citrate in healthy volunteers. Anesthesiology 92, 665–73.
150. Faull, C., McKechnie, E., Riley, J., and Ahmedzai, S. (1994). Experience with dipipanone elixir in the management of cancer related pain: case study. Palliative Medicine 8, 63–5.
151. Hoskin, P.J. and Hanks, G.W. (1991). Opioid agonist antagonist drugs in acute and chronic pain states. Drugs 41, 326–44.
152. Rance, M.J. (1979). Animal and molecular pharmacology of mixed agonist–antagonist analgesic drugs. British Journal of Clinical Pharmacology 7, 281–6.
153. Bullingham, R.E.S., McQuay, H.J., and Moore, R.A. (1983). Clinical pharmacokinetics of narcotic agonist-antagonist drugs. Clinical Pharmacology 8, 332–43.
154. Bullingham, R.E.S., McQuay, H.J., Dwyer, D., Allen, M.C., and Moore, R.A. (1981). Sublingual buprenorphine used post-operatively: clinical observations and preliminary pharmacokinetic analysis. British Journal of Clinical Pharmacology 12, 117–22.
155. Gal, T.J. (1989). Naloxone reversal of buprenorphine-induced respiratory depression. Clinical Pharmacology and Therapeutics 45, 6–71.
156. Mello, N.K. and Mendelson, J.H. (1980). Buprenorphine suppresses heroin use by heroin addicts. Science 207, 657–9.
157. Hammersley, R., Cassidy, M.T., and Oliver, J. (1995). Drugs associated with drug related deaths in Edinburgh and Glasgow, November 1990–October 1992. Addiction 90, 959–65.
158. Portenoy, R.K., Foley, K.M., and Inturrisi, C.E.
(1990). The nature of opioid responsiveness and its implications for
neuropathic pain: new hypotheses derived from studies of opioid
infusions. Pain 43, 273–86.
159. Neal, E.A., Meffin, P.J., Gregory, P.B., and Blaschke, T.F. (1979). Enhanced bioavailability and decreased clearance of analgesics in patients with cirrhosis. Gastroenterology 77, 96–102.
160. Giacomini, K.M., Giacomini, J.C., Gibson, T.P., and Levy, G.
(1980). Propoxyphene and norpropoxyphene plasma concentrations after
oral propoxyphene in cirrhotic patients with and without surgically
contructed portacaval shunt. Clinical Pharmacology and Therapeutics 28, 417–24.
161. Patwardhan, R.V. et al. (1981). Normal metabolism of morphine in cirrhosis. Gastroenterology 81, 1006–11.
162. Hasselstrom, J., Eriksson, L.S., Persson, A., Rane, A., Svensson, J., and Sawe, J. (1990). The metabolism and bioavailability of morphine in patients with severe liver cirrhosis. British Journal of Clinical Pharmacology 29, 289–97.
163. Chan, G.L. and Matzke, G.R. (1987). Effects of renal insufficiency on the pharmacokinetics and pharmacodynamics of opioid analgesics. Drug Intelligence in Clinical Pharmacology 21, 773–83.
164. McQuay, H.J. and Moore, R.A. (1997). Opioid problems, and morphine metabolism and excretion. In Handbook of Experimental Pharmacology (ed. A.H. Dickenson and J.-M. Besson), pp. 335–60. Berlin: Springer-Verlag.
165. Coyle, N., Adelhardt, J., Foley, K.M., and Portenoy, R.K. (1990). Character of terminal illness in the advanced cancer patient: pain and other symptoms during last four weeks of life. Journal of Pain and Symptom Management 5, 83–9.
166. Hanning, C.D. (1990). The rectal absorption of opioids. In Advances in Pain Research and Therapy Vol. 14 (ed. C. Benedetti, C.R. Chapman, and G. Giron), pp. 259–69. New York: Raven Press.
167. Coyle, N., Cherny, N.I., and Portenoy, R.K. (1994). Subcutaneous opioid infusions in the home. Oncology 8, 21–7.
168. Oliver, D.J. (1985). The use of the syringe driver in terminal care. British Journal of Clinical Pharmacology 20, 515–16.
169. Portenoy, R.K. (1987). Continuous intravenous infusions of opioid drugs. Medical Clinics of North America 71, 233–41.
170. Russell, P.S.B. (1979). Analgesia in terminal malignant disease. British Medical Journal i, 1561.
171. Bruera, E., Brenneis, C., Michaud, M., MacMillan, K., Hanson, J., and MacDonald, R.N.
(1988). Patient-controlled subcutaneous hydromorphone versus continuous
subcutaneous infusion for the treatment of cancer pain. Journal of the National Cancer Institute 80, 1152–4.
172. Waldmann, C.S., Eason, J.R., Rambohul, E., and Hanson, G.C.
(1984). Serum morphine levels. A comparison between continuous
subcutaneous infusions and intravenous infusions in post-operative
patients. Anaesthesia 39, 768–71.
173. O'Doherty, C.A., Hall, E.J., Schofield, L., and Zeppetella, G. (2001). Drugs and syringe drivers: a survey of adult specialist palliative care practice in the United Kingdom and Eire. Palliative Medicine 15, 149–54.
174. Grassby, P.F. and Hutchings, L. (1997). Drug combinations in syringe drivers: the compatibility and stability of diamorphine with cyclizine and haloperidol. Palliative Medicine 11, 217–24.
175. Moulin, D.E., Inturrisi, C.E., and Foley, K.M. (1986). Epidural and intrathecal opioids: cerebrospinal fluid and plasma pharmacokinetics in cancer pain patients. In Pain Research and Therapy Vol. 8 (ed. K.M. Foley and C.E. Inturrisi), pp. 369–84. New York: Raven Press.
176. Du Pen, S. and Williams, A.R. (1992). Management of patients receiving combined epidural morphine and bupivacaine for the treatment of cancer pain. Journal of Pain and Symptom Management 7, 125–7.
177. Hogan, Q., Haddox, J.D., Abram, S., Weissman, D., Taylor, M.L., and Janjan, N. (1991). Epidural opiates and local anesthetics for the management of cancer pain. Pain 46, 271–9.
178. Weinberg, D.S. et al. (1988). Sublingual absorption of selected opioid analgesics. Clinical Pharmacology and Therapeutics 44, 335–42.
179. Ripamonti, C. and Bruera, E. (1991). Rectal, buccal and sublingual narcotics for the management of cancer pain. Journal of Palliative Care 7, 30–5.
180. Zeppetella, G. (2001). Sublingual fentanyl citrate for cancer-related breakthrough pain: a pilot study. Palliative Medicine 15, 323–8.
181. Mercadante, S. et al. (2002). Episodic (breakthrough) pain. Cancer 94, 832–9.
182. Dale, O., Hjortkjaer, R., and Kharasch, E.D. (2002). Nasal administration of opioids for pain management in adults. Acta Anaesthesiologica Scandinavica 46, 759–70.
183. Kendall, J.M., Reeves, B.C., and Latter, V.S.
(2001). Multicentre randomised controlled trial of nasal diamorphine
for analgesia in children and teenagers with clinical fractures. British Medical Journal 322, 261–5.
184. Stein, C. (1995). The control of pain in peripheral tissues by opioids. New England Journal of Medicine 332, 1685–90.
185. Back, I.N. and Finlay, I. (1995). Analgesic effect of topical opioids on painful skin ulcers. Journal of Pain and Symptom Management 10, 493.
186. Krajnik, M. et al. (1999). Potential uses of topical opioids in palliative care—reports of 6 cases. Pain 80, 121–5.
187. Citron, M.L., Kalra, J.M., Seltzer, V.L., Chen, S., Hoffman, M., and Walczak, M.B. (1992). Patient-controlled analgesia for cancer pain: a long-term study of inpatient and outpatient use. Cancer Investigation 10, 335–41.
188. Walsh, T.D. and Cheater, F.M. (1983). Use of morphine for cancer pain. Pharmaceutical Journal 231, 525–8.
189. Brooks, D.J., Gamble, W., and Ahmedzai, S. (1995). A regional survey of opioid use by patients receiving specialist palliative care. Palliative Medicine 9, 229–38.
190. Ventafridda, V., Ripamonti, C., DeConno, F., Bianchi, M., Pazzuconi, F., and Panerai, A.E. (1987). Antidepressants increase bioavailability of morphine in cancer patients. Lancet i, 1204.
191. Cherny, N. et al. (2001). Strategies to manage the adverse effects of oral morphine: an evidence-based report. Journal of Clinical Oncology 19, 2542–54.
192. Donner, B. et al. (1996). Direct conversion from oral morphine to transdermal fentanyl: a multicenter study in patients with cancer pain. Pain 64, 527–34.
193. Babul, N., Provencher, L., Laberge, F., Harsanyi, Z., and Moulin, D. (1998). Comparative efficacy and safety of controlled-release morphine suppositories and tablets in cancer pain. Journal of Clinical Pharmacology 38, 74–81.
P.341
194. McDonald, P. et al. (1991). Regular subcutaneous bolus morphine via an indwelling cannula for pain from advanced cancer. Palliative Medicine 5, 323–9.
195. Baillie, S.P. et al. (1989). Age and the pharmacokinetics of morphine. Age and Ageing 18, 258–62.
196. Bentley, J.B. et al. (1982). Age and fentanyl pharmacokinetics. Anesthesia and Analgesia 61, 968–71.
197. Holdsworth, M.T. et al. (1994). Transdermal fentanyl disposition in elderly subjects. Gerontology 40, 32–7.
198. Rapin, C.H. (1989). The treatment of pain in the elderly patient. The use of oral morphine in the treatment of pain. Journal of Palliative Care 5, 54–5.
199. Osborne, R. et al. (1993). The pharmacokinetics of morphine and morphine glucuronides in kidney failure. Clinical Pharmacology and Therapeutics 54, 158–67.
200. Hagen, N.A. et al. (1991). Chronic nausea and morphine-6-glucuronide. Journal of Pain and Symptom Management 6, 125–8.
201. Sjogren, P. et al. (1993). Myoclonic spasms during treatment with high doses of intravenous morphine in renal failure. Acta Anaesthesiologica Scandinavica 37, 780–2.
202. Tiseo, P.J. et al. (1995). Morphine-6-glucuronide concentrations and opioid-related side effects: a survey in cancer patients. Pain 61, 47–54.
203. Bruera, E. et al. (1989). The cognitive effects of the administration of narcotic analgesics in patients with cancer pain. Pain 39, 13–16.
204. Fallon, M.T. and O'Neill, W.M.
(1998). Substitution of another opioid for morphine. Opioid toxicity
should be managed initially by decreasing the opioid dose. British Medical Journal 317, 81.
205. Sevarino, F.B.
et al. (1992). The efficacy of intramuscular ketorolac in combination
with intravenous PCA morphine for postoperative pain relief. Journal of Clinical Anesthesia 4, 285–8.
206. Bjorkman, R. et al. (1993). Morphine-sparing effect of diclofenac in cancer pain. European Journal of Clinical Pharmacology 44, 1–5.
207. Joishy, S.K. and Walsh, D.
(1998). The opioid-sparing effects of intravenous ketorolac as an
adjuvant analgesic in cancer pain: application in bone meta-stases and
the opioid bowel syndrome. Journal of Pain and Symptom Management 16, 334–9.
208. Minotti, V.
et al. (1998). Double-blind evaluation of short-term analgesic efficacy
of orally administered diclofenac, diclofenac plus codeine, and
diclofenac plus imipramine in chronic cancer pain. Pain 74, 133–7.
209. Portenoy, R.K. (1996). Adjuvant analgesic agents. Hematology/Oncology Clinics of North America 10, 103–19.
210. Queisser, W. (1984). Chemotherapy for the treatment of cancer pain. Recent Results in Cancer Research 89, 171–7.
211. Rubens, R.D. et al. (1992). Appropriate chemotherapy for palliating advanced cancer. British Medical Journal 304, 35–40.
212. Twycross, R.G. and Lack, S.A. Symptom Control in Far-Advanced Cancer: Pain Relief. London: Pitman Books, 1983.
213. Hanks, G.W. (1991). Opioid-responsive and opioid-non-responsive pain in cancer. British Medical Bulletin 47, 718–31.
214. de Stoutz, N.D. et al. (1995). Opioid rotation for toxicity reduction in terminal cancer patients. Journal of Pain and Symptom Management 10, 378–84.
215. Fallon, M. (1997). Opioid rotation: does it have a role? Palliative Medicine 11, 177–8.
216. Cherny, N.J.
et al. (1995). Opioid pharmacotherapy in the management of cancer pain:
a survey of strategies used by pain physicians for the selection of
analgesic drugs and routes of administration. Cancer 76, 1283–93.
217. Bruera, E.
et al. (1995). Changing pattern of agitated impaired mental status in
patients with advanced cancer: association with cognitive monitoring,
hydration, and opioid rotation. Journal of Pain and Symptom Management 10, 287–91.
218. Bruera, E.
et al. (1996). Opioid rotation in patients with cancer pain. A
retrospective comparison of dose ratios between methadone,
hydromorphone, and morphine. Cancer 78, 852–7.
219. Ashby, M.A. et al. (1999). Opioid substitution to reduce adverse effects in cancer pain management. Medical Journal of Australia 170, 68–71.
220. Pasternak, G.W. and Standifer, K.M.
(1995). Mapping of opioid receptors using antisense
oligodeoxynucleotides: correlating their molecular biology and
pharmacology. Trends in Pharmacological Sciences 16, 344–50.
221. Brosen, K. et al. (1993). Role of genetic polymorphism in psychopharmacology—an update. Psychopharmacology Series 10, 199–211.
222. Drexel, H. et al. (1989). Treatment of severe cancer pain by low-dose continuous subcutaneous morphine. Pain 36, 169–76.
223. Pies, R. (1996). Psychotropic medications and the oncology patient. Cancer Practice 4, 164–6.
224. Fallon, M. and O'Neill, B. (1997). ABC of palliative care. Constipation and diarrhoea. British Medical Journal 315, 1293–6.
225. Vainio, A. et al. (1995). Driving ability in cancer patients receiving long-term morphine analgesia. Lancet 346, 667–70.
226. Portenoy, R.K. (1994). Management of common opioid side effects during long-term therapy of cancer pain. Annals of the Academy of Medicine Singapore 23, 160–70.
227. Hanks, G.W., Twycross, R.G., and Lloyd, J.W.
(1981). Unexpected complication of successful nerve block.
Morphine-induced respiratory depression precipitated by removal of
severe pain. Anaesthesia 36, 37–9.
228. O'Neill, W.M., Hanks, G.W., Simpson, P., Fallon, M.T., Jenkins, E., and Wesnes, K.
(2000). The cognitive and psychomotor effects of morphine in healthy
subjects: a randomised controlled trial of repeated (four) oral doses
of dextropropoxyphene, morphine, lorazepam and placebo. Pain 85, 209–15.
229. Zacny, J.P. (1996). Should people taking opioids for medical reasons be allowed to work and drive? Addiction 91, 1581–4.
230. Warner, M.A. et al. (1991). Narcotic-induced histamine release: a comparison of morphine, oxymorphone, and fentanyl infusions. Journal of Cardiothoracic and Vascular Anesthesia 5, 481–4.
231. Katcher, J. and Walsh, D. (1999). Opioid-induced itching: morphine sulfate and hydromorphone hydrochloride. Journal of Pain and Symptom Management 17, 70–2.
232. McCaffery, M.
(1992). Pain control. Barriers to the use of available information.
World Health Organization Expert Committee on Cancer Pain Relief and
Active Supportive Care. Cancer 70 (Suppl. 5), 1438–49.
233. Ward, S.E. et al. (1993). Patient-related barriers to management of cancer pain. Pain 52, 319–24.
234. Mortimer, J.E. and Bartlett, N.L. (1997). Assessment of knowledge about cancer pain management by physicians in training. Journal of Pain and Symptom Management 14, 21–8.
235. Sees, K.L. and Clark, H.W. (1993). Opioid use in the treatment of chronic pain: assessment of addiction. Journal of Pain and Symptom Management 8, 257–64.
236. Schug, S.A. et al. (1992). A long-term survey of morphine in cancer pain patients. Journal of Pain and Symptom Management 7, 259–66.
237. Perry, S. and Heidrich, G. (1982). Management of pain during debridement: a survey of US burn units. Pain 13, 267–80.
238. Porter, J. and Jick, H. (1980). Addiction rare in patients treated with narcotics. New England Journal of Medicine 302, 123.
239. Medina, J.L. and Diamond, S. (1977). Drug dependency in patients with chronic headaches. Headache 17, 12–14.
240. Jasinski, D.R. (1981). Opiate withdrawal syndrome: acute and protracted aspects. Annals of the New York Academy of Sciences 362, 183–6.
241. Rogers, A.G. (1991). Prevention of the withdrawal syndrome in an opioid-dependent one-year-old child with decreasing pain. Journal of Pain and Symptom Management 6, 129.
242. Weissman, D.E. and Haddox, J.D. (1989). Opioid pseudoaddiction—an iatrogenic syndrome. Pain 36, 363–6.
243. Passik, S.D. et al. (1998). Substance abuse issues in cancer patients. Part 1: Prevalence and diagnosis. Oncology (Huntington) 12 (4), 517–21, 524.
244. Passik, S.D. et al. (1998). Substance abuse issues in cancer patients. Part 2: Evaluation and treatment. Oncology (Huntington) 12 (5), 729–34.
245. Passik, S.D. and Theobald, D.E. (2000). Managing addiction in advanced cancer patients. Why bother? Journal of Pain and Symptom Management 19, 229–34.